
اغلب بیماریهای نورولوژیک میتوانند علائم مختلف اختلالات حرکتی را ایجاد نمایند. دراین نوشتار اختلالات حرکتی مهم تر و شایعتر مورد بحث قرارگرفته است.
تشخیص اختلالات حرکتی به تجربه کافی ومشاهده دقیق بالینی نیازمند است. بدیهی است که گرفتن شرح حال دقیق اولین قدم درتشخیص است. اختلالات حرکتی علائم چشمگیروقابل مشاهده ای دارند واین ممکن است معاینه کننده را وسوسه نماید تا صرفاً براساس علائم مشهود ،درمورد تشخیص بیماری تصمیم گیری کند. باید تاکید نمود که شرح حال دقیق همانند سایررشته های طب بالینی الزامی است. بدون وجود شرح حال بالینی منا سب، اشتباه در تشخیص اجتناب ناپذیرخواهد بود. بعنوان مثال میتوان به اهمیت تاریخچه فامیلی در بیماری هانتیگتون اشاره نمود.همچنین در تشخیص فلج مغزی کودکان (cerebral palsy) شرح حال مربوط به وضعیت تکاملی و رشد کودک اهمیت خاصی دارد. سابقه مصرف داروهای نورولپتیک در بیماران مبتلا به پارکینسونیسم ویا دیس کینزی تاردیو اهمیت بسزایی دارد. حتی در یک بیماری بخصوص نحوه بروزعلائم بالینی همیشه به یک صورت نیست. مثلاً بیماری هانتینگتون ممکن است با هر یک از سندرمهای آکینتیک-رژید یا دیس کینتیک تظاهر نماید، یا یک بیمار مبتلا به بیماری پارکینسون که لوودوپا مصرف میکند ممکن است دچارانواع دیس کینزی شود.
اختلالات حرکتی شامل دو گروه اصلی هستند:
سندرمهای آکینتیک- رژید “Akinetic-rigid-syndromes” که با کندی حرکات و سفتی عضلا نی مشخص می شوند. به سندرمهای آکینتیک- رژید پارکینسونیسم نیزمیگویند. درسندرمهای آکینتیک- رژید پرسش اصلی این است که آیا بیمار به بیماری پارکینسون مبتلا است یا به دیگرسندرمهای آکینتیک- رژید دچار شده است.
۲ ) دیس کینزی ها “Dyskinesias” حرکات اضافه ای هستند که توسط خود بیماریااطرافیان اوبعنوان حرکتهای غیرطبیعی شناخته می شوند. اولین قدم در بررسی بیماران مبتلا به دیس کینزی مشخص نمودن نوع آن است. اغلب دیس کینزی ها در یکی از پنج گروه : ترمور، دیستونی، کره، میوکلونوس وتیک قرارمیگیرند.
پاتوفیزیولوژی اختلالات حرکتی
اختلالات حرکتی اغلب بعلت بیماری گانگلیون های بازال ایجاد می شوند. البته این مسئله در همه موارد صادق نیست. آن قسمت از گانگلیون های بازال که دراختلالات حرکتی اهمیت دارد خود از پنج هسته به شرح ذیل تشکیل شده است :
۱) پوتامن putamen
۲) هسته کودیت qaudate nucleus
یاد آوری میشود که به مجموعه پوتامن وهسته کودیت ،نئواستریاتوم neostriatum یا استریاتوم striatum میگویند.
۳) گلوبوس پالیدوس globus pallidus (GP) که به آن پالئواستریاتوم یا پالیدوم نیز گفته می شودو شامل دو بخش اکسترنال (GPe) واینترنال(GPi )است.
۴) ماده خاکستری substantia nigra (SN) که خود شامل دوبخش است.
– SN pars reticulata (SNr) که ازنظرتکاملی وعملکردهمانند GPi است.
-( ( SNc SN pars compacta که حاوی نورون های ترشح کننده دوپامین است.
۵) هسته ساب تالامیک subthalamic nucleus (STN).
اختلالات گانگلیونهای بازال معمولاً اثرات عمیقی بر سیستم حرکتی میگذارند و سبب بروز علائمی مانند تغییر درسرعت و دامنه حرکات ونیز اشکال درشروع و تداوم حرکات میشوند. این اختلالات شامل کلیه حرکات ارادی واتوماتیک، خودبخودی و رفلکسی می شوندو ازنظربالینی با کاهش وکندی حرکتی ویا بروزحرکت های اضافی غیر طبیعی تظاهرمی نمایند.
البته گانگلیون های بازال مرکزمنحصر به فرد کنترل حرکات نیستند و تنها بخشی از سیستم بسیار وسیع و پیچیده حرکتی را تشکیل می دهند. وجود ارتباطات وسیع گانگلیون های بازال با سیستم لیمبیک از یک طرف وبا فرآ یندهای شناختی از طرف دیگرنشانه عملکردهای متنوع این ارگان درسیستم عصبی می باشد. گانگلیون های بازال در پنج مدار عمده با نواحی مختلف کورتکس مغز درارتباط هستند. این پنج مدار نهایتاً سه بخش اصلی فانکشنال را در این سیستم تشکیل می دهند. این سه بخش به فعالیتهای شناختی، عاطفی وحرکتی گانگلیون های بازال مربوط می شوند.
نئواستریاتوم ایستگاه ورودی گانگلیون های بازال است و کلیه الیا ف تحریکی کورتیکواستریاتال را از نواحی مختلف کورتکس دریافت میکند. استریاتوم الیاف دوپامینرژیک نیگرواستریاتال را نیز از ماده خاکستری (SN) دریافت میکند. بخش حرکتی استریاتوم از طریق دو مسیر با گلوبوس پالیدوس اینترن یا GPi ارتباط دارد. مسیراولمسیرمستقیم و مسیر دوم مسیر غیرمستقیم نام دارد. در مسیر مستقیم ارتباط بدون واسطه ای بین استریاتوم و GPi وجود دارد. در حالیکه درمسیر غیرمستقیم الیاف استریاتو پالیدال ابتدا ازاستریاتوم به گلوبوس پالیدوس اکسترن( GPe )میروند و سپسازGPe به هسته ساب تالامیک( STN )و نهایتا به GPi وSNr مرتبط میشوند. GPi وSNrخروجی اصلی گانگلیون های بازال محسوب می شوند. در نهایت الیاف مهاری از GPiوSNr به تالاموس و سپس الیاف تحریکی از تالاموس به کورتکس میروند.


شکل-۱
با توجه به شکل وچگونگی ارتباط مسیرهای مستقیم و غیرمستقیم میتوان دید که افزایش فعالیت مسیر مستقیم سبب مهار خروجی گانگلیون های بازال و در نتیجه افزایش تحریک کورتکس حرکتی خواهد شد. در حالیکه افزایش فعالیت مسیر غیرمستقیم سبب افزایش فعالیت STN و در نتیجه افزایش فعالیت خروجی گانگلیون های بازال ونهایتاً کاهش تحریک کورتکس حرکتی میگردد. نوروترانسمیتردوپامین در مسیر مستقیم با عمل روی گیرنده های تیپ D1 دوپامینی با اثر تحریکی، و در مسیر غیرمستقیم با عمل روی گیرنده های تیپ D2 دوپامینی با اثر مهاری عمل می نماید.
در بیماری پارکینسون سلولهای سازنده دوپامین در ماده خاکستری از بین میروندو لذا تحریک دوپامینی از طریق راه نیگرواستریاتال کاهش می یابد.این کاهش سبب میشود که اثر طبیعی دوپامین یعنی تحریک مسیر مستقیم ومهار مسیرغیرمستقیم اتفاق نیافتد. در نتیجه الیف تالاموکورتیکال مهار شده و فعالیت کورتکس حرکتی کاهش می یابد. این مسئله سبب ایجاد علائم بیماری پارکینسون (که یک بیماری آکینتیک- رژید است) می شود.
هرگاه ابتدا در بخشهائی از استریاتوم که مسیرهای غیرمستقیم دران قرار دارند ضایعه ایجاد شود، بیماری با دیس کینزی تظاهر میکند (مانند شکل تیپیک بیماری هانتیگتون.) زیرا مهار یا تخریب مسیر غیرمستقیم سبب کاهش خروجی SNr-GPiوافزایش فعالیت مسیرهای تالاموکورتیکال می شود. با پیشرفت بیماری نواحی مربوط به مسیرهای مستقیم نیز آسیب می بینند و وضعیت دیس کینتیک بتدریج جای خود را به وضع آکینتیک- رژید میدهد.
در ضایعه هسته ساب تالامیک نیز( که علت درگیری آن معمولاً استروک است)در واقع مسیر غیرمستقیم آسیب شدید می بیند و در نتیجه یک وضعیت دیس کینتیک شدید با حرکات غیرارادی پرتابی دراندامهای طرف مقابل هسته درگیرایجاد می شودکه به آن همی بالیسم (hemiballism) میگویند.
پارکینسونیسم Parkinsonism
سندرمهای آکینتیک- رژید با کندی حرکات وسفتی عضلا نی مشخص می شوند. به این سندرمها پارکبنسونیسم نیزگفته می شود. مجموعاً ترکیبی ازشش علامت اصلی درپارکینسونیسم دیده می شود که عبارتند از
۱) ترمور در حال استراحت (tremor at rest)
۲) رژیدیته
۳) برادی کینزی
۴) اختلال رفلکس های وضعیتی (impairment-of-postural-reflexes)
۵) وضعیت خمیده به جلو (flexed-posture)
۶) جمود یا بلوک حرکتی (freezing or motor blocks)
مهمترین و شایعترین علت سندرمها ی آکینتیک- رژید بیماری پارکینسون است.
طبقه بندی سندرمهای آکینتیک- رژید:این سندروم ها درسه گروه عمده قرار میگیرند:
الف )پارکینسونیسم های اولیه بیماری های نورودژنراتیوی هستند که تظاهراصلی بیماری درآن ها بصورت سندرم پارکینسونیسم است وبیماریهای زیر راشامل میشود:
– بیماری پارکینسون
– سندرمهای پارکینسون Plus که شامل:
_مالتیپل سیستم آتروفی MSA
_فلج فوق هسته ای پیش رونده (Progressive Supranuclear Palsy) PSP
_دژنراسیون کورتیکوبازال CBD
ب)پارکینسونیسم های ثانویه یااکتسابی یا سمپتوماتیک که خودشامل موارد زیرمیباشد:
– پارکینسونیسم با علل دارویی:
داروهای بلوک کننده رسپتوردوپامین : داروهای آنتی سایکوتیک وضداستفراغ، رزرپین، تترابنازین، لیتیوم، فلوناریزین، سیناریزین
– پارکینسونیسم با علل عفونی:
بعد از آنسفالیت های ویروسی
– توکسین ها:
مونوکسید کربن، منگنز، مس، دی سولفید کربن، سیانید، متانول، اتانول و MPTP
– پارکینسونیسم عروقی:
– پارکینسونیسم متعاقب تروما ها
– سایرعلل پارکینسونیسم ثانویه:
اختلالات پاراتیرویید، هیپوتیروییدیسم، دژنراسیون هپاتوسربرال غیر ویلسونی، تومورهای مغزی، هیدروسفالی با فشار نرمال NPH
ج )پارکینسونیسم در سایراختلالات نورودژنراتیووارثی سندرم پارکینسونیسم دراین گروه علامت اصلی بیماری نیست وعلائم پارکینسونیسم درجریان بیماری اصلی ودر کنار سایرعلائم دیده می شود.دراین گروه بیماریهای زیز قرار میگیرند:
– بیماری هانتیگتون با توارث AD
– بیماری ویلسون با توارث AR
– بیماری هالروردن اشپاتزبا توارث AR
– کلسیفیکا سیون فامیلیال گانگلیون های بازال
بیماری پارکینسون (Parkinson’s disease)
یک بیماری نورودژنراتیو تدریجاً پیشرونده است ویکی از بیماری های شایع در نورولوژی بالینی محسوب می شود. از نظر بالینی وجود سه معیار برای تشخیص بیماری ضروری است.
۱)وجود دوعلامت ازسه علامت اصلی
– برادی کینزی یا کندی حرکتی
– رژیدیته و سفتی عضلانی
– ترمور در حال استراحت (tremor at rest).
۲)پاسخ بالینی به درمان بالوودوپا: لوودوپا در کلیه بیماران مبتلا به پارکینسون ودر کلیه مراحل بیماری مؤثراست وعدم پاسخ به درمان بالوودوپا با دوز کافی( در صورت لزوم تا ۵/۱ گرم روزانه )و مدت مناسب (تا سه ماه) تشخیص پارکینسون را زیرسوال می برد.
۳)عدم وجود علائمی که در بیماری پارکینسون معیار های آتیپیک محسوب می شوند شامل:
– درگیری سیستم پیرامیدال
– علائم درگیری مخچه ای
– افتالموپلژی و محدودیت حرکات چشم
– سقوط مکرردراوایل بیماری
– وجود آپراکسی
– د یسفاژی و دیزآرتری زودرس
– دیسفونی یا استریدور
– علائم زودرس و شدید درگیری اتونوم
– دمانس زودرس و وجود هالوسیناسیون های بینائی با مقادیر کم داروهای ضد پارکینسون
اپیدمیولوژی
شیوع پارکینسون در جمعیت های اروپایی در حدود ۲۵۰- ۱۰۰ مورد در یکصد هزار نفراست. در جمعیت پارسیان هند شیوع آن بیشتر بوده و در چینی ها وافریقایی ها کمتر شایع است. تفاوتی بین دو جنس وجود ندارد. سن بروز بیماری معمولاً پس از ۵۰ سالگی است و با افزایش سن شیوع آن بیشتر می شود. شروع بیماری قبل از ۴۰ سالگی young onset)) غیر معمول است و قبل از۲۰ سالگی نیز( juvenile ) بندرت اتفاق میافتد. در غیرسیگاریها شایعتراست که دلیل آن روشن نیست.
پاتوژنزوپاتولوژی
از نظر پاتولوژی بیماری پارکینسون با تخریب نورونهای پیگمانته دوپامینرژیک در ماده خاکستری( SN ) و وجود اجسام لِوی (Lewy bodies) درسلول های باقی مانده این ناحیه مشخص می شود. اجسم لوی را در سایر قسمت های ساقه مغز مثل لوکوس سرولوس (Locus-coeruleus) ، نوکلئوس دورسالیس واگوس و قسمت های دیگر سیستم عصبی مثل تالاموس، کورتکس مغز، نخاع و گانگلیون های سمپاتیک نیز میتوان دید. اجسام لوی انکلوزیون های داخل سلولی ائوزینوفیلیک هستند که حاوی ماده پروتیئنی alpha-synuclein می باشد. اجسام لوی دارای یک بخش مرکزی و یک هاله کمرنگ تر دراطراف هستند. وجود اجسام لوی منحصر به بیماری پارکینسون نیست و در بیماریهایی از قبیل آتاکسی تلا نژکتازی و بیماری هالروردن اشپاتز نیز دیده می شوند.
به نظر میرسدمکانیسم تخریب نورونهای پیگمانته ماده خاکستری ، عیب زنجیره تنفسی میتوکندریایی واسترس های اکسیداتیو ،قرار گرفتن در معرض مواد توکسیک و فاکتورهای ژنتیکی است و درنهایت برآ یند اثراین عوامل احتمال بروز بیماری را دریک فرد تعیین میکنند. توکسین های متنوعی در محیط و در صنایع مطرح شده اند ولی هنوز توکسین خاصی که سبب بیماری پارکینسون ایدیوپاتیک باشد پیدا نشده است. در سال ۱۹۸۳ ماده متیل- فنیل- تتراهیدروپیریدین MPTP شناخته شد که الودگی با آن سبب بیماری سریعاً پیشرونده با علائم شبیه بیماری پارکینسون ایدیوپاتیک می شد. امروزه از این ماده برای ایجاد بیماری پارکینسون درحیوانات آزمایشگاهی استفاده میکنند.
علائم بالینی بیماری پارکینسون
علائم بیماری بتدریج آغاز می شود واغلب تعیین زمان دقیق شروع بیماری ممکن نیست. برخی از بیماران از احساس خستگی و کندی شکایت میکنند. گاهی علائم به افسردگی یا کهولت سن نسبت داده می شود. دست خط بیمار تغییر میکند و ریز و بدمی شود. دردهای عضلا نی واحساس سفتی در عضلات بخصوص در عضلات اندام فوقانی و شانه شایع است. احساس درد واحساس لرزش درونی در عضو مبتلا، معمولاً اندام فوقانی شکایت غیر معمولی نیست. سپس بتدریج ترمور کلاسیک در حال استراحت، رژیدیته، و کندی حرکتی، یعنی علائم اصلی بالینی بیماری بارز می شود .
بیماری معمولاً بطور یکطرفه از اندامهای یک سمت و بطور تیپیک در یک دست شروع می شود. بندرت ممکن است بیماری از اندام تحتانی نیز شروع شود. حتی هنگامی که بیماری پیشرفت میکند واندامهای طرف مقابل را نیزدرگیرمیکند، این آسیمتریهمچنان باقی می ماند. بیمارازاختلال درحرکات دست یا کشیده شدن پا روی زمین شکایت میکند که تابلوی بالینی شبیه همی پارزی را بوجود می آورد ولی علامت بابنسکی وجود ندارد و رفلکس های تاندونی طبیعی هستند. هیچ تست تشخیصی پاراکلینیکی برای بیماری وجود ندارد وتشخیص منحصراً براساس شرح حال و معاینه بالینی باید داده شود. در مراحل اولیه و قبل از بارزشدن علائم اصلی که ممکن است ماهها بطول انجامد تشخیص بیماری آسان نیست واشتباهات تشخیصی بسیار شایع است.
همانطور که در مبحث پاتوفیزیولوژی اختلالات حرکتی گفته شد، اختلالات گانگلیونهای بازال سبب بروز علائمی مثل تغییردرسرعت و دامنه حرکات شده ومشکلاتی درشروع و تداوم حرکت ایجاد میکند. تغییر در سرعت حرکت در بیماری پارکینسون بصورت کندی حرکتی است که به آن برادی کینزی (bradykinesia) گفته می شود. دامنه حرکات نیز کاهش می یابد که به این حالت هیپو کینزی (hypokinesia)میگویند. به اختلال در شروع حرکت آکینزی (akinesia) گفته می شود. پد یده هایی مثل بلوک حرکتی و جمود حرکتی (freezing) نیزاز جمله اختلالات مربوط به شروع و تداوم حرکت هستند واز جمله علامت های آ کینتیک محسوب می شوند.
آ کینزی و هیپوکینزی و برادی کینزی مجموعاً در واقع مسبب بسیاری از تظاهراتعمده بالینی بیماری پارکینسون هستند. به مجموعه این سه علامت بطور کلی علائم آکینتیک نیز گفته شده است. صورت بی حالت و ماسکه، کاهش پلک زدن، کاهش حرکات طبیعی دستها در حال راه رفتن (arm-swinging) ، میکروگرافی، صدای کوتاه و مونوتون و راه رفتن کند با قدم های کوتاه از جمله این علائم می باشند. آکینزی مهمترین و ناتوان کننده ترین علامت بیماری پارکینسون است ومجموعاً یک فقر عمومی در حرکات خودبخودی ،اتوماتیک ، رفلکسی و نیزحرکات ارادی بیماران وجود دارد.
اشکال در تداوم حرکات، گاهی به صورت حالتی شبیه خستگی پذیری تظاهر میکند. دراین حالت، انجام حرکت های تکراری با کاهش پیشرونده دامنه حرکات توأم می شود تا سرانجام بیماراز ادامه حرکت باز می ماند. به این علا مت خستگی اکستراپیرامیدال (extrapyramidal fatigue) نیز گفته می شود.
رژیدیته عضلا نی (muscular rigidity)
بصورت مقاومت پاسیواعضاء در مقابل حرکت، توسط معاینه کننده حس می شود. معاینه کننده سفتی عضلا نی را بصورت مقاومت یکنواخت در تمام محدوده حرکتی گروه های عضلات آگونیست وانتاگونیست احساس میکند. به رژیدیته یکنواخت،رژیدیته لوله سربی یا lead pipe rigidity میگویند. هرگاه ترمور نیز همراه رژیدیته وجود داشته باشد، رژیدیته بصورت سفتی چرخ دندانه ای cog-wheel rigidity احساس می شود. وضعیت خمیده (flexed posture) نیز در بیماران بعلت رژیدیته عضلا نی ایجادمی شود.
ترموراستراحت (rest tremor)
شکایت اولیه دردوسوم بیماران است ودرنها یت دراکثربیماران ایجاد می شود. ترموردرحال استراحت با فرکانس ۶-۳ هرتز در تشخیص بیماری پارکینسون ارزش بسیاراختصاصی دارد و گاهی در دست شکل بسیار تیپیک تسبیح انداختن (pill-rolling)را پیدا میکند ترمور در پا و فک نیز ممکن است دیده شود. ترمور براثراسترس عاطفی تشدید میشود اما در خواب عمیق از بین می رود.
ناپایداری وضعیت (postural instability)
نیزازعلائم تیپیک پارکینسون بشمار می رود. بیماران در مراحل پیشرفته دچار علائم ناپایداری وضعیت می شوند که همراه با تمایل به سقوط به سمت عقب(retropulsion) است.
اختلال gait
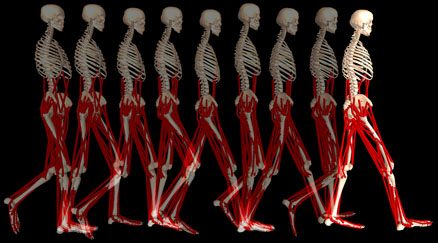
راه رفتن در بیماران پارکینسون بسیاراختصاصی است. حرکات طبیعی دست ها در حال راه رفتن (swinging arm movements) کاهش می یابدو مفاصل اصلی اندامهایعنی آرنج ، زانو و ران حالت فلکسیون خفیف پیدا میکنند. بیمار پاهای خود را روی زمین می کشد (shuffling) و با قدم های کوچک و کوتاه راه می رود بنابراین gait تیپیک در بیماران پارکینسونی راه رفتن آهسته ، در حالت خمیده وبا قدم های کوتاه همراه با کشیدن پاها روی زمین است.
زمانی که اختلال درپایداری وضعیت در مراحل پیشرفته بیماری، بصورت تمایل به سقوط رو به جلو(propulsion) باشد، تداخل این مشکل با اختلال راه رفتن در پارکینسون به پدیده نسبتاً کمیاب ولی بسیارکلاسیک festination منجر می شود. بیمار با هر قدمی که به جلو بر می دارد با احتمال سقوط به جلو مواجه است و برای اجتناب از سقوط، قدم بعدی را سریعتر برمی دارد و دراین حال قادربه توقف نیز نیست. بنابراین راه رفتن بیماربصورت یک راه رفتن شتاب گیرنده ،خمیده روبه جلوبا قدم های کوچک وسریع درمی آید. انگار که بیمار دنبال مرکز ثقل خود میدود. Festination نسبتاً کمیاب است.
در مراحل پیشرفته تراختلال gait بصورت اشکال درشروع راه رفتن بروز میکند(start hesitation) یا ممکن است که در حال راه رفتن دچار حالت انجماد حرکتیfreezing)) یا بلوک موتور نیز بشود. این حالت خصوصاً در فضاهای تنگ تر یا هنگام عبوراز درب اتاق ها یا در زمان ورود به محوطه های تنگ مثل آسانسور بیشتر بروز میکند. در نهایت بیمار ممکن است به مرحله ای برسد که بدون کمک قادر به راه رفتن نباشد. و بدون درمان قدرت هر حرکتی را از دست بدهد.
بیماران پار کینسونی را با توجه به شکل غالب علائم بالینی میتوان در نهایت در دو گروه اصلی قرار داد.
۱) گروه با علائم عمدتاً آکینتیک و رژید که درآن آکینزی و رژیدیته علائم غالب هستند.
۲) نوع tremor dominant که دران ترمور علامت اصلی وبارزاست.
پیش آگهی نوع دوم بهتراست. درمجموع تشخیص موارد تیپیک بیماری مشکل نیست. بیماررا با چهره بی حالت و ماسکه، وضعیت خمیده با ترمور rest و رژیدیته، کندی و فقر حرکتی، دست خط ریز و طرز راه رفتن تیپیک ، حتی یک غیر پزشک نیز میتواند باز شناسد.
سایر علائم و نشانه های بیماری پارکینسون
بجز علائم ونشانه های حرکتی مجموعه علائم دیگری نیز بدلایل مختلف در بیماران مبتلا به پارکینسون دیده می شود که یا بعلت خود بیماری ایجاد می شوند یا براثر عوارض جانبی داروها و یا بیماری های زمینه ای شایع از قبیل دیابت و هیپرتانسیون، یاصرفا بدلیل کهولت سن به وجود آمده اند.این علائم عبارتند از:
دمانس واختلالات منتال :دراین بیماران غیر شایع نیست. هر چنددراغلب موارد قوای شعوری و ذهنی دراین بیماری، حداقل در مراحل اولیه تغییر نمیکند، در بسیاری از بیماران با پیشرفت بیماری درجاتی از اختلال در توانائی های ذهنی بروز می نماید. شکل خاصی از کندی روند تفکر واختلال دریاد آوری موسوم به برادی فرنی (bradyphrenia)شایع است. شیوع دمانس در نتیجه مجموع عوامل در نهایت به حدود ۳۰ درصد می رسد و بیشتر به سن ارتباط دارد تا طول مدت بیماری. در مواردی که دمانس درارتباط مستقیم با پاتولوژی زمینه ای خود بیماری پارکینسون ایجاد شده باشد، در کورتکس مغز نیز لِوی بادی به مقدار قابل توجهی دیده میشود. هنگامی که علائم دمانس پس از یک سال از شروع بیماری پارکینسون مشهود گردد به آن پارکینسون بعلاوه دمانس(Parkinson’s disease with dementia) یا PDD گفته می شود.اما اگر فاصله بروز علائم دمانس از زمان شروع پارکینسون از یک سال کم تر باشد بیشتر دمانس بالوی بادی(dementia with Lewy bodies) یا DLB مطرح میشود. ازنظربالینی بیماران PDD و DLBممکن است خیلی شبیه به هم باشند. هرچند هالوسیناسیون های بینائی در هر دو شایع است اما اختلال توجه ، تمرکز،اختلال سطح هوشیاری و عملکرد شناختی تؤام با نوسان (fluctuation) مشخصه دمانس DLB است. ضمنا این بیماران دراثر یک اختلال سیستمیک یا عفونت ادراری یا تنفسی براحتی دچار دلیریم می شوند.
افسردگی :درحدود یک سوم بیماران پارکینسونی از افسردگی رنج می برند. دراکثر موارد شدید نیست و میتواند از نوع راکتیو باشد. تشخیص ودرمان به موقع افسردگی در بهبود کیفیت زندگی این بیماران مهم است. بیماری پارکینسون ممکن است اشتباهاً بعنوان افسردگی تشخیص داده شود والبته عکس آن نیز اتفاق می افتد.
شکایت های حسی: علیرغم معاینه حسی طبیعی در بیماری پارکینسونشکایات حسی شایع است. بخصوص درد اندامها که بطور شایعی دیده میشود. گاهی آکاتژیا (Akathisia) یا بی قراری حرکتی در پاها بروز میکند. احساس درد یا کرخی دراندامهای مبتلا ممکن است از علائم اولیه باشد و شک به مشکلات عضلا نی اسکلتی را ایجاد کند، از قبیل بورسیت یا جمود مفصل شانه یا دردهای سیاتیکی. اگردرد ژنرالیزه باشد علائم به بیماری های رماتیسمی یا بیماری پلی میالژی رُماتیکا شباهت پیدا میکند. در مجموع تابلوی بالینی دردهای اندام و کندی حرکتی ممکن است به راحتی به حساب کهولت سن بیمار گذاشته شود و بیماری پارکینسون تشخیص داده نشود.
گاهی دفورمیته های ظاهری اسکلتی در دست ها و پاها شبیه آرتریت روماتویید دیده می شود. علت اغلب این موارد وجود رژیدیته است و با مصرف دارو به میزان کافی بر طرف می شود.
استئوپورز در گروه سنی مبتلایان به پارکینسون شایع است و شکستگی های استخوانی بدنبال سقوط مکرر دراین بیماران فراوان دیده میشود.
خستگی : یک علامت شایع است وممکن است ژنرالیزه یا محدود به یک اندام باشد. خستگی ممکن است درابتدا محدود به یک فعالیت خاص مثل نوشتن باشد ومثلاً با کرامپ نویسندگان اشتباه شود.
علائم چشمی : بصورت بلفاروکلونوس (انقباضات ریتمیک عضلات پلک) و بلفاروسپاسم، گاهی دیده می شود. فلج convergence ومحدودیت نگاه(gaze limitation) به بالا نیز دیده می شود. حرکات ساکادیک و تعقیبی (pursuit) چشم ها در معاینه بالینی نرمال است.
اختلالات گوارشی :یبوست وکاهش وزن بسیار شایع است. ریزش بزاق از دهان (drooling) ود یسفاژی در بسیاری از بیماران درمراحل پیشرفته بیماری دیده می شود.
اختلالات ادراری: ناشی ازبیماری پارکینسون بصورت تحریک پذیری مثانه همراه با تکررادرار، urgency و urge incontinence است. در مردان بزرگی پروستات نیز بر مشگلات فوق می افزاید.
افت فشار خون وضعیتی(postural hypotension) :ممکن است در نتیجه در گیری سیستم اتونوم با پاتولوژی لوی بادی دیده شود. علت شایع آن مصرف داروهای دوپامینرژیک است.
باید توجه داشت که گرچه دراکثر موارداین گونه علائم با وجود بیماری پارکینسون قابل توجیه هستند، ولی توصیه می شود به هر علامت بعنوان مشکل بالینی خاص توجه شده واز نظر رد علل احتمالی بررسی های لازم انجام شود.
بررسی های تشخیصی
MRI مغزدربیماری پارکینسون ایدیوپاتیک نرمال است.Single-Photon-Emission-Computerized-Tomography (SPECT) میتواند اختلال درسیستم دوپا مینرژیک گانگلیون های بازال را نشان بدهد ولی در تشخیص افتراقی سندرمهای مختلف آکینتیک- رژیدازیکدیگرکمکی نمیکند.SPECT به تشخیص افتراقی پارکینسون با تابلوی غالب ترمور،ازبیماری ترموراسانسیل کمک میکند. این بررسی درترموراسانسیل وپارکینسونیسمدارویی نرمال است.
درمان
بهتر است درمان بیماری در اولین فرصت ممکن با توجه به شرایط فردی بیمار شروع شود. در صورت تاخیر در درمان ایجاد پلاستیسیته منفی در مغز میتواند پاسخ بیمار را به درمان دچار اشکال نماید.
شش نوع دارو دردرمان علامتی پارکینسون استفاده می شود:
-آنتی کلینرژیک های سنترال
-آمانتادین
-ترکیبات لوودوپا
-مهار کننده های مونوآمین اکسیداز MAOI-B
-مهار کننده های کاتکول اُ متیل ترانسفراز COMT
-آگونیست های دوپامین DAs
در درمان بیماران سمپتوماتیک نوع داروی مصرفی را باید باتوجه به سن و وضعیت منتال بیماران انتخاب کرد. بهترین رژیم درمانی برای شروع درمان در بیماران جوانتر آگونیست های دوپامین است که در صورت نیاز می توان لوودوپا را به آن اضافه نمود. بهتر است در بیماران مسن درمان با لوودوپا شروع شود و تا حد ممکن از داروهای دیگر استفاده نشود.
ترکیبات آنتی- کلینرژیک ضد پارکینسون رایج درایران عبارتند از دو داروی آنتی کلینرژیک سنترال بنام تری هگزی فنیدیل و بی پریدن. این دارو ها دارو های ضد پارکینسون ضعیفی محسوب می شوند و به ترتیب پس از لوودوپا وآگونیست های دوپامین وآمانتادین از نظراثرات ضد پارکینسونی در مرتبه آخرقرار میگیرند. آنتی کلینرژیک ها روی مهمترین و ناتوان کننده ترین علامت پارکینسون یعنی آکینزی و برادی کینزی اثری ندارند و روی ترمور و تاحدی رژیدیته مؤثرهستند. گفته شده است درصورتی که بیماری خفیف وعلامت اصلی آن تنها ترمور باشد میتوان از آنتی کلینرژیک ها استفاده کرد. به اشتباه تصور می شود که آنتی کلینرژیک ها درمان انتخابی ترمورهستند. طبق دستورالعمل های منتشر شده در تشخیص و در مان پارکینسون، نباید از آنتی کلینرژیک ها در شروع درمان پارکینسون استفاده شود. موثر ترین دارو در کنترل ترمور وسایر علائم پارکینسون لوودوپا است. دوز مؤثر لوودوپا در بیماران مختلف برای کنترل ترمور متغییراست و دربرخی موارد ، مقدارداروی مورد نیاز برای کنترل ترمور بیشترازمقادیری است که برای کنترل سایر علائم لازم میشود. ترموراز نظر فانکشنال معمولاً مشکلی برای بیمارایجاد نمیکند ولی بسیاری از بیماران روی کنترل ترموراز نظر ظاهری اصرار دارند .
با توجه به سیر طبیعی بیماری واثرات عمیقی که مجموعه علائم بیماری بالقوه میتواند روی روند زندگی بیمار داشته باشد باید برای بیمار روشن نمود که کنترل ترمور به تنهائی هدف درمان محسوب نمی شود. درمراحل اولیه معمولاً استراتژی درمان براساس بهبود عملکرد و فانکشن اندامها بخصوص عملکرد دست تعیین می شود، خصوصاً اگر بیماری در دست غالب شروع شده باشد. در مراحل پیشرفته تر بیماری بهتر شدن پایداری وضعیت (postural-stability) وراه رفتن بیمار، هدف اصلی درمان را تشکیل می دهد. هدف حذف کامل کلیه علائم بیماری نیست و منظوراز درمان برقراری امکان فعالیت مناسب روزمره برای بیمارمتناسب با نیازهای اصلی زندگی است. یک نکته دیگر نیزتصور نادرست از نقش آنتی کلینرژیک ها در کنترل علائم پارکینسون را تقویت میکند. قطع ناگهانی یا سریع داروهای آنتی کلینرژیک موجب افزایش حاد و شدید کلیه علائم پارکینسون می شود واین مسئله گاهی اشتباهاً به حساب مؤثربودن این ترکیبات گذاشته می شود. داروهای آنتی کلینرژیک را بخصوص در بیماران با علائم پیشرفته باید بسیارآهسته وبه تدریج قطع نمود. اثر داروهای آنتی کلینرژیک در کنترل علائم بیماری اندک است وعوارض محیطی (خشکی دهان، تاری دید، یبوستواحتباس ادراری) وعوارض سنترال (اختلال حافظه، تغییرات شخصیتی و دلیریوم) ایجاد میکنند. این عوارض بسیار شایع هستند. وبیماران پارکینسونی بخصوص بیماران مسن به دلیل خود بیماری، بالقوه در معرض اکثراختلالات مذکور هستند. این دارو ها را نباید درافراد مسن مصرف کرد. با توجه به گروه سنی مبتلایان به بیماری پارکینسون، میتوان نتیجه گرفت که دارو های آنتی کلینرژیک عملاً در درمان اکثر بیماران مبتلا به پارکینسون جایگاهی ندارند.
آمانتادین یک داروی ضد ویروس است که اثرات مثبت آن در درمان پارکینسون تصادفاً شناخته شده است. مکانیسم اثرآن شامل جلوگیری از جذب مجدد دوپامین، اثرشبه آمفتامینی تخلیه دوپامین، اثرانتی کلینرژیک واثر روی گیرنده های اِن- متیل- د-آسپارتاتNMDA می باشد. دارو با مکانیسم اخیر روی دیس کینزی های ناشی از دوپامین مؤثراست.
ترکیبات لوودوپا (levodopa) مؤثرترین دارو روی کلیه علائم پارکینسون و در واقعgold-standard درمان این بیماری محسوب می شوند. چون خود دوپامین نمیتواند از سد خونی- مغزی عبور نماید از ماده پیش ساز آن یعنی لوودوپا استفاده میکنند که در مغز به دوپامین تبدیل می شود. برای اینکه لوودوپا قبل از ورود به CNS به دوپامین تبدیل نشود آن را بصورت ترکیب با مهار کننده های محیطی دوپادکربوکسیلاز مثل کربی- دوپا(carbidopa) و بن سرازید (benserazide) بکار می برند. بهتراست برای پیشگیری از عوارض اتونوم و گوارشی درمان را با دوزکم، ۱۰۰-۵۰ میلی گرم لوودوپا روزانه یکبار پس از غذا شروع نماییم سپس بتدریج مقدار دارو را به میزان لازم افزایش داده و به آرامیوعده های مصرف دارو را نیز به قبل از غذا منتقل کنیم. غذا بخصوص غذا های پروتئینی مانع جذب لوودوپا می شوند.
وقتیکه هنوز بیماری پیشرفت نکرده است و درمان بالوودوپا شروع می شود بیماران پاسخ یکنواخت و مناسب به لوودوپا می دهند و درفاصله دوزهای داروو حتی در صورت عدم مصرف یک یا چند نوبت دارو ، در علائم حرکتی نوسانی را احساس نمیکنند. باگذشت زمان و با پیشرفت بیماری اثر یکنواخت و بدون نوسان لوودوپا به اثر وابسته به دوزتبدیل می شود و در صورت تاخیر در مصرف هر نوبت دارو علائم پارکینسون خودنمائی میکند. به این حالت، خاتمه یافتن اثر دارو”wearing-off” یا بدتر شدن علائم درانتهای اثر دوز دارو “end of dose deterioration” گفته می شود. به این ترتیب بیمار دوره های به اصطلاح روشن و خاموش “on-off” را تجربه میکند که زمان بروز آن قابل پیش بینیمیباشد چراکه با دفعات مصرف و زمان اثر دارو و مشخصاً با خاتمه و شروع اثر دارو ارتباط مستقیم دارد.
علاوه براین موارد، به مرور پدیده حرکات اضافی غیرارادی نیز در برخی از بیماران به مشکلات درمانی افزوده می شود. به این حرکت های اضافی که در بیماران با مصرف لوودوپا بروز میکند، دیس کینزی های ناشی از لوودوپا “Dopa induced Dyskinesias” (DID) میگویند. مشخص نیست که این عوارض حرکتی که در جریان درمان بالوودوپا در بیماران پارکینسونی اتفاق می افتد آیا در نتیجه عوارض لوودوپا است یا دراثر پیشرفت بیماری ایجاد می شود.
عوارض روانی لوودوپا: بصورت دلیریوم، هالوسیناسیون های (معمولاً )بینائی و دوره های سایکوز دیده می شود.
عوارض اتونوم : عارضه شایع لوودوپا هیپوتانسیون ارتواستاتیک است.
هیچ کنتراندیکاسیون مطلقی برای درمان بالوودوپا وجود ندارد. لوودوپا را نباید همراه یا به فاصله کمتراز ۱۵ روز از قطع داروهای مهار کننده مونوآمین اکسیداز A(MAOI-A) تجویز نمود.فراموش نکنیم که لوودوپا در بیمارانی که اخیراً دچارانفارکتوس میوکارد شده اند نباید مصرف شودو اصولا بهتراست از حداقل دوز لازم در هر بیماراستفاده گردد.
سلژلین selegiline یک داروی مهار کننده مونوآمین اکسیداز B (MAOI-B) است که اثر کمی روی علائم پارکینسون دارد وتا حدی نیز میتواند در برخی از بیماران مدت اثر لوودوپا را افزایش بدهد. متابولیت آمفتامینی دارد. اثر نوروپروتکتیوان هنوز درانسان ثابت نشده است.
آگونیست های مستقیم دوپامین را میتوان هم از ابتدای درمان و هم برای کنترل مشکلات درمانی تاخیری ،همراه با لوودوپا بکار برد. این داروها دو دسته هستند.داروهای bromocriptine ، pergolide ، lisuride و cabergoline مشتفاتergot و داروهای ropinirole و pramipexole و apomorphine ترکیبات non-ergot محسوب می شوند. هرچند هیچ یک از این دارو ها در کنترل علائم پارکینسون به اندازه لوودوپا مؤثر نیستند ولی اگر در شروع درمان از آنها استفاده شودعوارضی از قبیل پدیده های نوسان در پاسخ درمانی on-off))و دیس کینزی بندرت اتفاق می افتد. در هر حال دیر یا زود اثر درمانی آگونیست ها کفایت نخواهد کرد واستفاده از لوودوپا اجتناب ناپذیر خواهد بود.
درمانهای غیر دارویی
دراکثر بیماران برنامه فعالیت منظم روزانه، فیزیوتراپی، گفتار درمانی و مشاوره با متخصصین تغذیه( برای تعیین رژیم مناسب غذائی) بسیار کمک کننده است.
روش های جراحی در بیماری پارکینسون
با پیدا شدن لوودوپا در سالهای ۶۰ میلادی و تاثیر فوق العده آن در کنترل علائم بیماری درمانهای جراحی که قبل از آن برای کنترل علائم بیماری رواج یافته بود به کناری نهاده شد. ولی با توجه به مشکلات درمانی و نوسانات در پاسخ به درمان که گاه تنظیممقدار مصرف آن را بسیار دشوار می سازد و نیز ایجاد دیس کینزی های مربوط به درمان بالوودوپا، رویکرد مجدد به روش های جراحی ،بخصوص در دهه گذشته با پیشرفت و موفقیت های چشمگیری در کنترل علائم بیماری همراه بوده است. در گذشته و برای سال های طولانی رایج ترین روش جراحی تالاموتومی یکطرفه بود. تالاموتومی به کنترل ترمور و تا حدی رژیدیته کمک میکند ولی تاثیری روی آکینزی ندارد. تالاموتومی دو طرفه سبب اختلال تکلم، حافظه و راه رفتن می شود.
پالیدوتومی در کنترل دیس- کینزی های ناشی از لوودوپا دراندامهای سمت مقابل وتا حدی دراندامهای همان سمت مؤثراست و علائم پارکینسونیسم را نیز دراندامهای سمت مقابل در حدود ۳۰-۲۵ درصد کاهش می دهد. اثر پالیدوتومی روی ترمور کم است. پارکینسونیسم دراندامهای همان سمت تغییر چندانی نمیکند. این روشروی اختلالات راه رفتن، postural-instability واختلال تکلم نیز تاثیری ندارد. عوارض جراحی پالیدوتومی عبارتند از خونریزی، همی پارزی، اختلال میدان بینائی.
درمقابل روش های تالاموتومی و پالیدوتومی که روش های تخریبی (با ایجاد ضایعه) محسوب می شوند، روش جدیدتری به نام تحریک عمقی مغز (Deep-Brain-Stimulation) رواج یافته است. در این روش الکترودی در هدف مورد نظر در مغز قرار داده می شود و سیم آن به یک استیمولاتور زیر جلدی متصل می گردد. روش هایDBS از نظر تکنیکی دشوارتر و وقت گیر تراز روش های تخریبی هستند. اثر DBS تالاموس و DBS پالیدال از نظر کنترل علائم شبیه تالاموتومی و پالیدوتومی است. نتایج روش جدیدتر DBS در هسته های زیر تالاموس (Subthalamic nucleus) یا STN-DBS بسیار چشمگیر بوده است. در بیماران در مراحل پیشرفته پارکینسون که دچار نوسانات موتور و دیس کینزی هستند STN-DBS در کنترل این عوارض وسایر علائم پارکینسون و حتی کنترل اختلال gait موفق بوده است. بدون مصرف دارو STN-DBS به تنهائی ۶۰ درصد علائم بیماری را کاهش می دهد ونیاز بیمار به مصرف دارو به کمتراز ۵۰ درصد می رسد. اضافه میکنیم که درمان با پیوند بافت و پیوند سلول های جنینیدرحال حاضر روش های تحقیقاتی محسوب می شوند.
سندروم های پارکینسون پلاس
۱-فلج فوق هسته ای پیشرونده (Progressive Supranuclear Palsy)
PSP بیماری نورودژنراتیو پیشرونده و نادری است که شیوع آن در حدود ۶ در ۱۰۰۰۰۰ است. بیماری اسپورادیک بوده و بین ۴۵ تا ۷۵ سالگی با سن متوسط ۶۳ سال بروز میکند. طول عمر بیماران پس از شروع علائم بین ۷-۵ سال است. آکینزی و رژیدیته اَکسیال (axial rigidity) گاهی همراه با دیستونی سرویکال بصورت کشیده شدن سر به سمت عقب (retrocollis)، postural instability واختلال gait بارزو زودرسبصورت retropulsion همراه با سقوط مکرر، اختلال پیشرونده بلع و تکلم بعلت فلج پسودوبولبار، فلج فوق هسته ای حرکات چشم و دمانس تیپ فرونتال از علائم بیماری هستند. در این بیماری که شیوع بیشتری در مردان دارد، حرکات ساکادیک چشم ها در گیر می شود. ابتدا در نگاه رو به پایین محدودیت ایجاد می شود و سپس حرکت چشم ها در سایر جهات نیز محدود می شود. با وجود در گیری حرکت های ساکادیک ارادی،رفلکس های اکولوسفالیک واکولو وستیبولر تا اواخر بیماری نسبتاً سالم می مانند. بلفاروسپاسم نیزدر برخی بیماران دیده می شود.(رجوع شود به بخش نشانه شناسی)علائم در گیری پیرامیدال مثل افزایش رفلکس ها و علامت بابنسکی و علائم در گیری مخچه ای نیز گاهی دیده می شود. این بیماران برخلاف بیماران پارکینسونی وضعیت خمیده ندارند.
MRI در مراحل پیشرفته آتروفی midbrain را نشان می دهد. تغییرات پاتولوژیکبصورت آتروفی و گلیوز همراه با کلافه های نوروفیبلیری (neurofibrillary tangle) در ساقه مغز، گانگلیون های بازال و به میزان متغیری در کورتکس مغزمیباشد. از نظر درمانی فقط در حدود ۲۰ درصد از بیماران به آمانتادین پاسخ می دهند. برای تخفیف دیستونی گردن و بلفاروسپاسم می توان از تزریق توکسین بوتولیسم استفاده نمود.
۲-مالتیپل سیستم آتروفی (Multiple System Atrophy)
یماری نورودژنراتیواسپورادیک در بالغین است. شیوع آن ۴ در ۱۰۰۰۰۰ است و بین ۳۳ تا ۷۶ سالگی با سن متوسط ۵۳ سال بروزمیکند. طول عمر بیماران از زمان شروع بیماری بین ۵ تا ۱۰ سال است. از نظر بالینی MSA میتواند با مجموعه علائم مخچه ای، پارکینسونیسم، اتونوم و پیرامیدال بروز نماید. درپاتولوژی علائم مرگ سلولی و گلیوز در نواحی ماده خاکستری، استریاتوم ، هسته های زیتونی olive و هسته دورسال واگوس،پونز، مخچه و ستون های intermediolateral نخاع و هسته Onufs در قسمت ساکرال نخاع دیده می شود. مشخصه پاتولوژی MSAوجود انکلوزیون های داخل سیتوپلاسمیدرسلولهای اولیگودندروگلیال است این انکلوزیون ها حاوی ماذه پروتئینی بنام alpha-synuclein می باشند.
نحوه تظاهر بالینی MSA به نسبت در گیری سیستم های استریاتونیگرالواولیوپونتوسربلار بستگی دارد. در صورتی که در گیری در سیستم استریاتونیگرال بیشتر باشد علائم پارکینسونیسم بارز بوده، به آن دژنراسیون استریانو نیگرال یا MSA-P گفته می شود. این فرم بصورت یک سندرم آکینتیک- رژید پیشرونده تظاهر میکند. در مراحل اولیه یک سوم بیماران پاسخ خوب و یک سوم دیگر نیز پاسخ نسبی به درمان بالوودوپا می دهند. یک سوم باقیمانده نیز به لوودوپا پاسخ نمی دهند. با گذشت زمان کلیه بیماران پاسخ اولیه به لوودوپا را از دست خواهند داد و میتوان گفت که پس از پنج سال از شروع بیماری دیگر هیچ یک از بیماران MSA-P به لوودوپا پاسخ نمی دهد. علائمMSA-P بر خلاف پارکینسون بیشتر بصورت سیمتریک شروع می شود. ترموراغلب بصورت ترمورpostural است.
درفرم اولیوپونتوسربلار یا MSA-C بیماری عمدتاً بصورت آتاکسی اسپورادیک پیشرونده تظاهر میکند و با پیشرفت بیماری بتدریج علائم آکینتیک- رژید نیز به آن اضافهمی شود.
نارسائی اتونوم در هر دو فرم بیماری دیده می شود واگر تابلوی بالینی عمدتاً بصورت نارسائی اتونوم باشد به آن سندرم شای- دراگر Shy-Dragerیا MSA-A گفته می شود. علائم ا تونوم در بیماران اغلب بصورت ناتوانی جنسی در مردان واختلال اسفنکتری است. هیپوتانسیون وضعیتی ( postural )و کاهش تعریق نیز شایع است. مواردی که با نارسائی ا تونوم شروع می شوند در مدت پنج سال سایر تظاهرات نورولوژیک بیماری را نیز نشان می دهند.
استریدور و دیسفونی ، اختلالات خواب، اختلالات رفتاری مربوط به خواب مرحلهREM و antecollis (خمیدگی بیش از حدّ گردن به جلو) از جمله علائم نسبتاً شایع و مهم MSA هستند. تشخیص با شرح حال و معاینه بالینی داده می شود.
در MRI ممکن است آتروفی مخچه، آتروفی پوتامن همراه با تغییر سیگنال در حاشیه خارجی آن و تغییر سیگنال در فیبرهای متقاطع پونز شبیه علامت بعلاوه دیده شود.
پس از آشنایی با چند نمونه از سندروم های رژید- آکینتیک ،حال به شرح چندنمونه از انواع دیس کینزی میپردازیم:
دژنراسیون کورتیکوبازال (Corticobasal degeneration)
بیماری بسیار نادراسپورادیک در بالغین است. شروع بیماری معمولاً بعد از ۶۰ سالگی است بصورت پارکینسونیسم بروز میکند که با آکینزی، رژیدیته، میوکلونوس، آپراکسی، از بین رفتن حس های کورتیکال، دیستونی شدید و یک طرفه دراندام فوقانی، و پدیده اندام بیگانه (alien limb) بدلیل درگیری supplementary motor area مشخص میگردد. postural instability و سقوط مکرر ممکن است به سرعت به تابلوی بالینی اضافه شود. در یک سوم موارد ترموردر حال استراحت rest دیده می شود. ترمور با ترمور تیپیک پارکینسونی متفاوت است. ترمور سریعتر بوده، غیر منظم است و همراه با آن میوکلونوس های فوکال دیده می شود. درادامه دمانس واختلال حرکات چشم به صورت اختلال فوق هسته ای حرکتی چشم ها وعلائم پیرامیدال نیزایجاد می شود. بندرت علائم از اندام تحتانی ویا بصورت دو طرفه شروع می شود. گاهی علائم دمانس از ابتدای شروع بیماری وجود دارد. گاهی آفازی نیز دیده می شود. تشخیص این بیماری با شرح حال و معاینه بالینی است. MRI مغز اغلب آتروفی آسیمتریک فرونتال و پاریتال را نشان می دهد. از نظر پاتولوژیک مرگ نورونی وایجاد نورون های آکروماتیک بالونی شکل در کورتکس نواحی فرونتال و پاریتال وگانگلیون های بازال وجه مشخصه بیماری است. درمان مؤثری وجود ندارد و طول عمر بیماران از زمان شروع علائم ۷-۵ سال است.
ترمور Tremor
ترمور یک حرکت ریتمیک سینوزوئیدی ومتناوب است. محل شایع بروز ترمور در دست ها است ولی در تنه، سر، عضلات صورت، و پاها نیز دیده می شود. پس از مشخص شدن شرح حال باید ترمور را براساس شکل ظاهری آن و رابطه اش با وضعیت و فعالیت عضو درگیر طبقه بندی نمود. علیرغم تعاریف و تقسیم بندی های مختلفی که موجود است، از نظر بالینی تقسیم ترمور به سه نوع rest, action, intention کاربرد بیشتری دارد.
ترمور rest درزمانی که عضو مبتلا بدون حرکت روی تکیه گاه قرار داشته باشد و یا هنگامی که عضو به حالت آویخته قرار گیرد بارزتر می شود. مانند ترمور rest در بیماران پارکینسونی ، هنگام قرار دادن دست ها روی تکیه گاه، ویا هنگامی که بیماربا دست های آویخته راه می رود.
ترمور action/postural درزمان حرکت یا فعالیت ارادی و یا حفظ ارادی وضعیت عضو در مقابل نیروی جاذبه ایجاد می شود. مانندبلند کردن دستها ونگه داشتن آنها دراین وضعیت
ترمور intention با نزدیک شدن عضوبه هدف در یک حرکت هدفمند (goal-directed) ایجاد می شود. مثلاً در تست finger to nose.
طبقه بندی ترمور
ترمور rest
بیماری پارکینسون
سندرمهای آکینتیک- رژید
ترمور action/postural
ترمور فیزیولوژیک
ترمور فیزیولوژیک تشدید شده
تیروتوکسیکوز
اختلالات اضطرابی
قطع الکل
داروها (سمپاتومیمتیک ها، لیتیوم، سدیم والپروآت)
ترموراسانسیل ET
ترمور intention
ضایعات مخچه ای وساقه مغزی
مالتیپل اسکلروز
بیماری عروقی
تومورها
حال به ذکر چند نوع ترمور شایع میپردازیم :
۱-ترمور نرمال (فیزیولوژیک) Physiologic tremor
ترمور فیزیولوژیک در همه اشخاص وجود دارد که با فرکانس ۱۰- ۸ هرتزدر دستهای به جلو کشیده قابل مشاهده وثبت است ولی معمولاً بدون علامت است. این ترمور با تحریک آدرنرژیک ، اضطراب ، مصرف داروهای سمپاتومیمتیک وقهوه، هیپوگلیسمی وتیرتوکسیکوزتشدید می شود. داروهائی مثل والپروات سدیم ولیتیوم نیز سبب تشدید ترمور فیزیولوژیک می شوند. برای کنترل ترمور فیزیولوژیک تشدید شده در صورت نیاز میتوان از بتا بلوکر ها مثل پروپرانولول استفاده نمود.
۲-ترموراسانسیل خوش خیم Benign essential tremor
ترموراسانسیل ET به میزان قابل توجهی ازبیماری پارکینسون شایعتراست. بیماری از نظر زمان شروع دارای دو پیک سنی است. یکی در ۱۵ سالگی و بعدی در ۵۰ سالگی. دربیش از نیمی از موارد سابقه فامیلی با توارث اتوزومال غالب وجود دارد. هیچ آنومالی پاتولوژیک یا بیوشیمی درتعداد اندکی که مورد نکروپسی قرار گرفته اند مشاهده نشده.
در مواردی که ترمور وضعیتی (postural) خفیف تا متوسط بدون سابقه خانوادگی وجود دارد تشخیص بین ترموراسانسیل و ترمور فیزیولوژیک تشدید شده(Enhanced-physiologic-tremor) بسیار مشکل می شود. فرکانس ET معمولا کمتراز ترمور فیزیولوژیک است و در حدود ۸ – ۶ هرتز است.( اما باید بدانیم که اندازه گیری فرکانس با توجه به وجود مواردی که فرکانس همسانی دارند روش مطمئنی برای تشخیص محسوب نمی شود.) شایعترین محل بروز ترموراسانسیل اندامهای فوقانی است. دریک سوم موارد دراندامهای تحتانی نیز دیده می شود. در موارد شدید، ترمور در گردن، زبان، صدا، عضلات فک وصورت نیزبروزمیکند. وجود ترمور ایزوله در هر یک از نواحی مذکور به نفع تشخیص ET نیست.ET ، معمولاً با بیماری نورولوژیک دیگری همراهی ندارد، اما اخیرا مطرح شده است که وقوع اختلالات خفیف شناختی واختلالات راه رفتن بصورت Unsteady gait یا آتاکسی gait دراین بیماری محتمل تراست. در مواردی نیزترمورهای شبیه ET درنوروپاتی محیطی یا در دژنراسیون های مخچه ای دیده می شود.
در برخی از بیماران مبتلا به پارکینسون ،ترمور postural شبیه به ترموراسانسیل دیده می شود اما درترموراسانسیل علائم پارکینسونیسم وجود ندارد. قابل ذکراست که گاهی ممکن است حالت چرخ دنده ای (Cog-wheeling) بدون رژیدیته در ترمور اسانسیلدیده شود.
بیماری دراکثر موارد سیر بسیار کند دارد و عمدتاً تنها سبب مشکل ظاهریواجتماعی برای بیمار می شود. البته افرادی که کارشان به حد بالائی از مهارت دستینیاز دارد ممکن است در نتیجه این بیماری متحمل دشواری فراوان شوند.
مقدار کم یا متوسط الکل معمولاً بطورقابل توجه ترموررا کاهش می دهد .از نظر درمانی در حدود نیمی از بیماران به درمان با بتا بلوکرها مثل پروپرانولول، پاسخ نسبتاً قابل قبولی می دهند. پریمیدون در برخی از موارد مفید واقع می شود. داروهای ضدپارکینسون اثری روی ET ندارند. در موارد شدید، تحریک عمقی DBS)) یکطرفه یا دوطرفه تالاموس میتواند بسیار موثرباشد.
ترمور مخچه ای Cerebellar (intention) tremor
ترموریکی از علائم مهم در بیماری های مخچه است که همراه دیس متری و سایر علائم درگیری مخچه دیده می شود. بیماری های مخچه سبب بروز ترمور در حال حرکت دادن اندامها می شوند وهنگامی که اندام در مسیر حرکت به هدف تعیین شده نزدیکمی شود ترمور تشدید می شود. به همین دلیل این ترمور را ترمور intention نامگذاری کرده اند. ترمور intention را با تستهای finger to nose و heel to shin میتوان ارزیابی نمود. این ترمور( بجز در موارد بسیار خفیف) معمولاً با دیس متری همراه است. مشخصه بسیار مهم ترمور های مخچه ای اینست که عمدتاً عضلات محوری و پروگزیمال را درگیر میکنند. آسیب های ناحیه میانی و ورمیس مخچه سبب آتاکسی gait و titubation سر وتنه می شود در صورتی که ضایعات نیمکره های مخچه یا مسیرهای خروجی مخچه سبب اتاکسی دراندامها و بروز ترمور intentional می شود.متاسفانه درمان خاصی برای ترمور مخچه ای یا intention tremor وجود ندارد.
ترمور پارکینسونی
رجوع شود به مبحث علائم بالینی بیماری پارکینسون.
کره Chorea
کره chorea حرکات سریع، مداوم، غیرارادی، نامنظم و بی هدف اندامها است که با الگوی غیر قابل پیش بینی از یک قسمت بدن به قسمت دیگر جریان پیدا میکند. اغلب در نواحی دیستال اندامهاو با دامنه کم بروز میکند.( حرکات پروگزیمال با دامنه بالا همی بالیسم نامیده می شود.) بسیاری اوقات بیمارممکن است حرکت کره ای را درمتن یک حرکت دلخواه ارادی پنهان کند. در هم کشیدن چهره (facial grimacing) ،صدا های غیر طبیعی تنفسی و هیپوتونی نیز در کره شایع است.
طبقه بندی کره
بیماری های دژنراتیووارثی
بیماری هانتیگتون
کره ارثی خوش خیم
آتاکسی تلا نژکتازی
کره های سمپتوماتیک واسپورادیک
داروها
آنتی کلینرژیک ها
نورولپتیک ها
فنی توئین ، کاربامازپین
ضد افسردگی های تری سیکلیک
سایرعلل کره
کره سیدنهام
کره گراویداروم (کره دوران بارداری)
کره بعلت مصرف قرصهای ضد بارداری
کره متعاقب عفونت استرپتوککی با آنتی بادی بر علیه گانگلیون های بازال
لوپوس و سندرم آنتی فسفولیپید
تیروتوکسیکوز
هیپوپاراتیروئیسم
هیپرگلیسمی
هیپرناترمی
پلی سیتمی ورا
بیماری های عروق مغز/تومورهای مغز/ ترومای مغز
کره سیدنهام Sydenham’s chorea
گاهی متعاقب تب روماتیسمی (Rheumatic fever)یا عفونتهای استرپتوکوکی گروه Aدر کودکان ،کره وبرخی اختلالات حرکتی دیگربروز مینماید. این نوع کره که امروز نسبتا کمیاب تر از گذشته دیده میشود ،به کره سیدنهام یاSt Vitus dance معروف است. بیماری کره سیدنهام که در دختر بچه ها شایعتر است گاهی همراه با اختلالات رفتاری و روانپزشکی نیز همراه میشود، و دربرخی مواردبا آنتی بادی های ضد گانگلیونهای بازال همراه میشود.
شروع کره بصورت تحت حاد است و میتواند با هیپوتونی و ضعف عضلا نی همراه باشد. دراغلب موارد تغییرات منتال بارزتوام با علائم رفتاری و عاطفی دیده می شود. کره میتواند یکطرفه باشد وحتی در بعضی از بیماران بصورت همی بالیسم تظاهر نماید. EEGگاهی تغییراتی بصورت امواج آهسته را نشان می دهد و درMRI مغزگاهی تورم وتغییر سیگنال در گانگلیوتهای بازال قابل مشاهده است. بیماری معمولاً پس از یک دوره ۶-۳ ماهه بتدریج بهبود میابد.
در مرحله حاد برای کنترل علائم میتوان از هالوپریدول استفاده کرد. آنتی کلینرژیک ها سبب تشدید علائم کره می شوند و نباید در کنار هالوپریدول از داروهای آنتی کلینرژیک استفاده شود. والپروآت سدیم نیز در کنترل کره مؤثراست و بهتراست درمان را ابتدا با والپروآت سدیم شروع نماییم.
بیماری هانتیگتون Huntington’s disease
هانتیگتون بیماری نادرارثی با توارث اتوزومال غالب است. یک بیماری نورودژنراتیو پیشرونده است که معمولا در حوالی میانسالی شروع می شود و با مجموعه علائم اختلالات حرکتی ، (معمولاً بصورت کره )، اختلالات رفتاری و اختلالات شناختی مشخص میشود. در تمام نواحی دنیا دیده می شودو شیوع آن در حدود ۱۰-۵ در یکصد هزار جمعیت است. اتیولوژی بیماری موتاسیون ژنی بصورت تکرار تری نوکلئوتید CAG روی بازوی کوتاه کروموزوم ۴ میباشد. هرچه طول قطعه موتاسین یافته تکرار تری نوکلئوتید CAGبیشتر باشد بیماری در سن کمتر و با شدت بیشتر بروز میکند. این حالت معمولاً هنگامی اتفاق می افتد که ژن معیوب از پدر منتقل شده باشد.
شکل تیپیک بیماری بین سنین ۳۰ تا ۵۰ سالگی دیده می شود. ابتدا علائم رفتاری بصورت تغییرات شخصیتی وعاطفی بروز میکند. افسردگی، کاهش انگیزه و کم عمق شدن عواطف بیمار برای اطرافیان مشخص میگردد. تحریک پذیری وبدخلقی، رفتار کنترل نشده تهاجمی یا رفتار غیر معمول جنسی بروز مینماید. اختلال حرکتی بصورت کره تظاهر میکند. با پیشرفت بیماری ابتدا به شدت کره افزوده شده و سپس بتدریج علائم سندرم آکینتیک- رژید ودیستونی به آن اضافه می شود و گاهی حتی به تابلوی بالینی غالب تبدیل میگردند. اختلال حرکات چشم واختلال بلع وتکلم نیز از علائم مهم بیماری هستندو نهایتاً بیمار کاملاً ناتوان وبیحرکت می شود. سیربیماری از شروع علائم بطور متوسط ۱۵ سال است.
از نظر پاتولوژیک آتروفی ژنرالیزه مغز همراه با در گیری بارز در گانگلیونهای بازال در ناحیه استریاتوم (هسته های کودیت وپوتامن) دیده می شود.
متاسفانه درمانی برای بیماری وجود ندارد. معمولاً برای کنترل کره داروهای بلوک کننده دوپامین مانند هالوپریدول تجویز می شود. بنظرمی رسد تجویز این داروها بجز کنترل ظاهری کره ، نه تنها هیچ نفعی برای بیماران ندارد بلکه با توجه به عوارض جانبی قابل توجه و سیر بعدی علائم بیماری احتمالاً پیش آگهی را برای آنان بدترنیز میکند. استفاده از داروهای selective serotonin reuptake inhibitors-SSRI وداروهای آنتی سایکوتیک آتیپیک جدید برای این بیماران در کنترل علائم توصیه می شود.
بررسی ژنتیکی به جهت تشخیص بیماری وشناسائی بیماری قبل از بروز علائم درافرادی که والدین شان مبتلا به بیماری هستند امکانپذیراست.
همی بالیسم Hemiballism
به حرکات پرتابی اندامهای فوقانی و تحتانی در یک طرف همی بالیسم گفته می شود. این حرکت ها شبیه کره است ولی بطور عمده عضلات پروگزیمال اندامها وکمربند شانه ولگن در گیر میکند. همی بالیسم بیماری نادری است. معمولاً درافراد مسن مبتلا به دیابت و هیپرتانسیون، در نتیجه وقوع استروک دیده می شودو دراین صورت علائم بطور ناگهانی بروز میکنند.
شدت همی بالیسم در بیماران مختلف متفاوت است و در موارد شدید میتواند سبب آسیب دیدگی بیمار شود. همی بالیسم ثانوی به استروک معمولاً بتدریج در مدت چند ماه بهتر می شود. در صورت لزوم برای تخفیف علائم میتوان از داروهای نورولپتیکneuroleptics یا تترابنازین (tetrabenazine) استفاده نمود.
میوکلونوس Myoclonus
میوکلونوس انقباضات کوتاه و شوک مانند عضلا نی منظم یا غیر منظم در یک عضله یا گروهی از عضلات است. اگرمیوکلونوس فقط دریک ناحیه تکرار شود به آن میوکلونوس فوکال میگویندواگر چند ناحیه آناتومیک مجاورهم را در گیر کرده باشد میوکلونوس سگمنتال نامیده می شود. اگر در زمان های متفاوت در قسمتهای مختلف بروز کند به آن میوکلونوس مولتی فوکال میگویند واگر بطور یکپارچه در سراسر بدن بروز کند میوکلونوس ژنرالیزه است.
طبقه بندی میوکلونوس
براساس مشخصات بالینی
Aنحوه ایجاد
Action
Spontaneous
Reflex
Bنحوه توزیع
ژنرالیزه
مولتی فوکال
سگمنتال
فوکال
براساس منشاء نوروفیزیولوژیک
کورتیکال
ساب کورتیکال ساقه مغز
اسپاینال
براساس اتیولوژی
Aمیوکلونوس فیزیولوژیک
Bمیوکلونوس اسانسیل
Cمیوکلونوس سمپتوماتیک
aهمراه اپی لپسی
bهمراه با سایربیماری ها
طبقه بندی سندرمی میوکلونوس
آتاکسی های میوکلونیک پیشرونده (PMA) progressive myoclonic ataxia
بیماری میتوکوندریال MERRF
دژنراسیون های اسپینوسربلار
بیماری سلیاک
اپی لپسی های میوکلونیک پیشرونده (PME) progressive myoclonic epilepsy
بیماری میتو کوندریال MERRF
بیماری لافورا Lafora*
سندرم *Unverricht-Lundborg
بیماری تای- ساکس
سروئید لیپوفوشینوزیس
سیالیدوز
آنسفالوپاتی های میوکلونیک پیشرونده
بیماری لافورا Lafora*
سندرم *Unverricht-Lundborg
دمانس های میوکلونیک (myoclonic dementias)
پان آنسفالیت اسکلروزان تحت حادSSPE
بیماری کروتزفلد- جاکوب
بیماری الزهایمر
دژنراسیون کورتیکو بازال
اتیولوژی های مختلف ممکن است با سندرمهای متفاوت تظاهر نمایند.*
میوکلونوس ژنرالیزه و مولتی فوکال Generalized and multifocalmyoclonus
در انواع مختلف بیماریهای نورولوژیک وانسفالوپاتی های متابولیک یا توکسیک دیده می شود. در بسیاری موارد میوکلونوس در نتیجه دیس شارژهای کورتیکال بطور خودبخودی یا در پاسخ به محرک خارجی اتفاق می افتد. این میوکلونوس های کورتیکال قرابت زیادی با اپی لپسی دارند.
میوکلونوس اپی لپتیک ممکن است علامتی از یک اپی لپسی ژنرالیزه(Myoclonic Epilepsy )یا یک بیماری پیشرونده نورولوژیک باشد.
میوکلونوس ممکن است تابلوی غالب یک بیماری باشد. در این موارد میوکلونوس بطور خودبخودی یا بدنبال حرکت (Action myoclonus)و یا در پاسخ به محرک های بینایی صوتی ویا سوماتوسنسوری (Reflex myoclonus) ایجاد شود. میوکلونوس حرکتی بعد از آنوکسی مغزی (Post-anoxic action myoclonus) دیده می شود.
هرچندمیوکلونوس ممکن است در برخی از دمانس های دژنراتیو مثل بیماری آلزهایمر دیده شوداما از علائم اختصاصی بیماری کروتزفلد- جاکوب (Creutzfeldt-Jakob)بشمار می رود.
میوکلونوس های فوکال
در برخی بیماریها میوکلونوس فقط در یک ناحیه از بدن دیده می شود. میوکلونوس فوکال می تواند در نتیجه دیس شارژ های عصبی در تمام سطوح سیستم اعصاب ایجاد شود. در کورتکس ، ساقه مغز، نخاع ، و در اعصاب محیطی.
همی فاسیال اسپاسم نوع بسیارشایعترمیوکلونوس فوکال است. بیماری با انقباضات ظریف و سریع غیر منظم از گوشه خارجی یک چشم شروع می شود وبتدریجسایر عضلات نیمه صورت را نیز در گیر می نماید. گاهی انقباضات بصورت اسپاسم های طولانی تر سبب جمع شدن غضلات دور چشم و بالا کشیده شدن دهان می شود. در اغلب موارد ضعف خفیف عضلات صورت در سمت مبتلا وجود دارد. علت شایع آن ضایعه عصب فاسیال در نزدیکی محل خروج عصب از ساقه مغز است. که توسط یک رگ خونی تحت فشار قرار می گیرد. اغلب بیماران به تزریق توکسین بوتولینوم یا مصرف کاربامازپین پاسخ مطلوب می دهند.
درمان: اغلب میوکلونوس ها به درمان با داروهایی مانند والپروات سدیم، کلونازپام، پیریمیدون، پیراستام و Levetiracetam (Keppra) پاسخ میدهند.
تیک Tic
تیک ها حرکات تکراری هستند که با الگوی معین (بصورت استرئوتایپ) در یک قسمت از بدن، بطور عمده در صورت، سر وقسمت بالای تنه دیده می شوند. ولی بیمار میتواند بطورارادی برای مدت محدودی مانع وقوع آن شود. پلک زدن مکرر، چین انداختن روی بینی، تکان دادن شانه همراه با کج کردن گردن از تیک های ساده شایع هستند.
تیک ممکن است حرکتی یا صوتی باشد. تیک های حرکتی را به انواع ساده و کمپلکس تقسیم میکنند. تیک های ساده بصورت حرکت های ساده سریع در یک گروهازعضلات اتفاق می افتند.تیک های حرکتی کمپلکس، حرکت های پیچیده هماهنگی هستند که معمولاً چند گروه عضله درانجام آن دخالت میکنند، مثل دستکاری یا بوکردن اشیاء و بدن، نشان دادن حالت های مختلف چهره، خاراندن ولگد زدن. ظاهرهماهنگ و پیچیده این حرکت ها تمیز آن ها را ازحرکات ارادی دشوارمی سازد. در بسیاری موارد تیک با حرکات compulsive تؤام است. که این امر تشخیص ماهیت حرکت را بعنوان تیک دشوارتر میکند. . هرگاه تیک حرکتی ظاهر آهسته وکند داشته باشد و بصورت یک انقباض مداوم انجام شود، مثل چرخاندن گردن یا انقباض عضلات پلک که شبیهبلفاروسپاسم باشد، به آن تیک دیستونیک میگویند.
تیک های صوتی نیز ممکن است ساده یا کمپلکس باشند. تیک های صوتیساده شامل صاف کردن گلو، سرفه، ملچ ملچ کردن، پارس کردن، خرناس کشیدن وایجاد صدا های ساده و تکراری است. تیک های صوتی کمپلکس بیشتر بصورت بیانکلمات یا جملات خاصی است که بیمار بدون دلیل ظاهری ناگهان آن را ادا میکند و ممکن است یک کلمه عادی ویا زشت و بی ادبانه باشد یا حتی محتوای مذهبی داشته باشد.
ازبیماریهایی که تیک تابلوی بالینی بارز آنها را تشکیل میدهد به سندروم ژیل دولاتوره اشاره میکنیم.
سندرم ژیل دولا توره Gilles de la Tourette syndrome
علائم بیماری قبل از ۱۸ سالگی شروع می شود. بنظر میرسد که یک اختلال ارثی باشد و احتمالاً الگوی توارث آن اتوزومال غالب است. تیک ها بیشتردر قسمتهای بالای تنه بخصوص صورت، گردن و شانه ها ظاهر می شوند. شدت و نحوه توزیع تیک در طول زمان متغییر است و دوره های بهبودی نسبی و تشدید علائم در سیر بیماری وجود دارد. الگوی تیک نیز متغیر است و شکل تیک ها در دوره های زمانی مختلف متفاوت است. اغلب این بیماران دیر یا زود دچار تیک های صوتی نیز می شوند، مثل خرناس کشیدن، فریاد زدن، صاف کردن گلو یا سینه، سرفه و پارس کردن. در حدود ۱۵ درصد بیماران ،صداها به کلمات زننده (Coprolalia) تبدیل می شود.
در بسیاری از بیماران علائم اختلال وسواسی- جبری (Obsessive-compulsive disorder) OCD وجود دارد. اختلال توجه و تمرکز همراه با بیش فعالی (Attention-deficit hyperactivity disorder) ADHD در کودکان مبتلا به تورت شایع است.
بیماری معمولاً در تمام عمر ادامه پیدا میکند. البته معمولاً در بزرگسالی از شدت علائم کاسته می شود. تیک ها وصدا ها میتوانندفشارروانی زیادی برای بیمار ایجاد کنند.
درمان : داروهای آنتی دوپامینرژیک مانند Pimozide، Sulpiride، هالوپریدول و تترابنازین ممکن است به کنترل تیک ها و صداهای اضافی کمک کند. داروها باید با دوز کم شروع شود و مقدار آن بتدریج متناسب با پاسخ بالینی تنظیم گردد. بیماران تورت در مقابل عوارض اکستراپیرامیدال نورولپتیک ها از اشخاص عادی مقاوم ترند و نیازی به استفاده همزمان آنتی کلینرژیک ندارند. برای علائم OCD دارو های مهار کننده انتخابی جذب مجدد سروتونین Serotonin reuptake inhibitors (SSRIs) و جهت ADHD متیل فنیدات توصیه می شود.
دیستونی Dystonia
دیستونی یا اسپاسم های عضلانی مداوم وغیر منظم تکرارشونده هستند که اغلب سبب تغییر شکل غیرعادی متغیر وگاهی مداوم دراندامها یا تنه می شوند. برای مثال دیستونی در گردن بصورت چرخش گردن به یک سمت (تورتیکولی)، خم شدن گردن به یک طرف ، کشیده شدن سر به عقب ، و یا خم شدن گردن به جلو دیده می شود. دیستونی در تنه یا دیستونی ترانکال، معمولاً به لوردوز یا کیفواسکولیوز منجر می شود. دراندام فوقانی اکستانسیون و هیپرپروناسیون بازو وفلکسیون مچ دست واکستانسیون انگشتان دیده می شود. اسپاسم های دیستونیک دراندامهای تحتانی معمولاً با ایجاد اکستانسیون، فلکسیون و چرخش بداخل مچ پا سبب تغییر وضعیت (posture) طبیعی پامی شود. dorsiflexion شست پا معروف به “striatal toe” شایع است.
درابتدا اسپاسم های دیستونیک ممکن است فقط با انجام کارهای بخصوصی ایجاد شوند. مثلاً دیستونی دراندام تحتانی فقط هنگام راه رفتن ایجاد می شود. و یا تغییردیستونیک وضعیت دست فقط هنگام نوشتن دیده می شود. به این حالت “action dystonia” یا “task-specific dystonia” گفته می شود. با پیشرفت بیماری اسپاسم های غیر طبیعی عضلانی و تغییر وضعیت اندامها در حال استراحت و بطور خودبخود نیزایجاد می شود.
دیستونی ممکن است فقط در یک ناحیه از بدن بروز کند، مثل دیستونی سرویکال (تورتیکولی اسپاسمودیک)، که دراین صورت به آن دیستونی موضعی یا دیستونی فوکال میگویند. هرگاه دیستونی دو ناحیه آنانومیک مجاور را درگیر نماید، مثل دیستونی سرویکال همراه با دیستونی دراندام فوقانی به آن دیستونی سگمنتال میگویند. دیستونی دراندامهای فوقانی و تحتانی یک سمت را همی دیستونی می نامند. و هرگاه دیستونی در تمام نواحی باشد به آن دیستونی ژنرالیزه گفته می شود.
همچنین دیستونی ها را به دو گروه عمده اولیه و ثانویه تقسیم می کنند.
در دیستونی های اولیه یا idiopathic torsion dystonia معمولاً دیستونی تنها علامت بالینی است و سایر علائم درگیری نورولوژیک دیده نمی شود. در بسیاری از بیماران سابقه خانوادگی مثبت است واغلب نحوه توارث اتوزومال غالب را نشان می دهند. هیچ یافته اختصاصی پاتولوژیک دردیستونی های اولیه ژنرالیزه مشاهده نشده است.
دیستونی های ثانویه یا سمپتوماتیک در نتیجه بیماری های نورولوژیک مختلف ایجاد می شوند. و معمولاً بجز دیستونی سایرعلائم درگیری سیستم عصبی نیز قابل مشاهده است.
دیستونی های ژنرالیزه یا سگمنتال اولیه
شایعترین نوع دیستونی ژنرالیزه و سگمنتال در نتیجه deletion ژن DYT1 روی کروموزوم ۹ ابجاد می شود. نفوذ ژن در حدود ۴۰-۳۰ درصد است بنابراین تاریخچه فامیلی تعداد قابل توجهی از بیماران منفی است.
بیماری معمولا در کودکی آغاز می شود. ومعمولاً با اسپاسم های دیستونیک هنگام راه رفتن دراندامهای تحتانی بروز میکند. گاهی دیستونی از دست، تنه یا گردن شروعمیشود. راه رفتن کودک بتدریج تغییر میکند ویا دچاراشکال در نوشتن می شود. بیماری هرگاه در کودکی شروع شود معمولاً سیر پیشرونده داردواسپاسم های دیستونیک از محل اولیه بتدریج به نواحی دیگرانتشارمی یابند. انتشاراسپاسم های دیستونیک بعد از بلوغ متوقف می شود. درافرادی که از نظر ژنتیکی مبتلا هستند، هرگاه بیماری تا سن ۲۶ سالگی شروع نشده باشد، بروز علائم بعد از این سن بعید است. معمولاً توانایی های ذهنی طبیعی است و علائم درگیری پیرامیدال واختلال حسی مشاهده نمی شود.
درمان دیستونی اولیه ژنرالیزه بسیاردشواراست. درحدود ۵ درصدازبیماران به درمان با لوودوپا پاسخ دراماتیک می دهندک䝇 درگروه دیستونی با پاسخ به لوودوپا(DRD) قرارمیگیرند. هرگاه پاسخ به لوودوپا منفی باشد، موفقترین دارو در مرحله بعد، آنتی کلینرژیک ها هستند که معمولاً از تری هگزی فنیدیل استفاده می شود. دارو با مقدار کم شروع می شود و درطول چندین ماه بتدریج به مقادیر بالا میرسد تا حدی که پاسخ بالینی ایجاد شود یا اینکه بعلت عوارض جانبی امکان افزایش بیشتر دارو ممکن نباشد. در حدود ۵۰ درصد بیماران به درمان با تری هگزی فنیدیل پاسخ می دهند.
چنانچه بیماربه هیچ یک از دو داروی لوودوپا یا تری هگزی فنیدیل پاسخ ندهد، احتمال پاسخ به سایر درمانهای دارویی بسیاراندک خواهد بود. با این وجود برخی از بیماران به درمان با باکلوفن خوراکی، کاربامازپین، یا یک بنزودیازپین تا حدی پاسخ می دهند. باکلوفن تزریقی اینتراتکال در تعدادی از بیماران با دیستونی بسیار شدید مؤثر بوده است. باکلوفن اینتراتکال بویژه درموارد شدید فلج مغزی آتتویید (Athetoid cerebral palsy) که با فلج اسپاستیک نیز همراه باشد بسیار مؤثر بوده است. اخیراً نتایج خوبی از درمان جراحی تحریک عمقی مغز (deep brain stimulation) DBS پالیدال دوطرفه بدست آمده است.
دیستونی های فوکال اولیه
دیستونی هایی که در سنین بالاتر شروع می شوند از نظر ژنتیک کمتر شناخته شدهاند. شیوع ژن DYT1 دراین گروه از بیماران بسیار کم است ولی در عده کمی از موارد فامیلیال ژن های دیگری شناخته شده است. اغلب دیستونی های اولیه فوکال اسپورادیک و غیرارثی هستند و یا ممکن است در نتیجه موتاسیون های جدید ایجاد شده باشند.
تورتیکولی اسپاسمودیک
تورتیکولی اسپاسمودیک شایعترین فرم دیستونی های فوکال محسوب می شود. در زنان از مردان شایعتراست. ومعمولاً در میاتسالی یا دیرتر بروز میکند. شروع علائم تدریجی است. درد از اولین علائم وشایع است. گاهی دیستونی بدنبال ترومای گردن ایجاد می شود. همانطور که قبلاً گفته شد به چرخش سر به یک سمت تورتیکولیس گفته می شود. خم شدن سر به جلوانته کولیس، خم شدن سر به عقب رتروکولیس و خم شدن سر به سمت شانه لاتروکولیس نامیده می شود. اسپاسم ها ممکن است تکرار شونده باشند و درگیری عضلات آنتاگونیست بطور همزمان ممکن است سبب لرزش سر شود (dystonic head tremor).
در حدود یک هشتم بیماران remission خودبخودی دیده می شود ولی معمولاً موقتی است. در معاینه عصبی بیماران بجز تورتیکولی، مورد دیگری ندارند واز سایر جهات طبیعی هستند. در برخی موارد ترمور postural شبیه ترموراسانسیل دیده می شود.
درمان دارویی معمولاً بی اثراست یا اثر بسیار کمی دارد. درمان انتخابی تزریق توکسین بوتولیسم به عضلات مبتلا است و دراغلب بیماران سبب کنترل قابل توجه دیستونی می شود. تزریقات معمولاً بفواصل هر سه ماه یکبار تکرار می شود. تعداد کمی از بیماران از ابتدا نسبت به اثر توکسین مقاوم هستند (مقاومت اولیه). و برخی دیگر در طول درمان پاسخ مطلوب اولیه را بتدریج بعلت تولید آنتی بادی های خنثی کننده توکسین از دست می دهند (مقاومت ثانویه). درافراد با مقاومت ثانویه عمل جراحیselective partial neurectomy را میتوان توصیه نمود. عمل جراحی DBS دوطرفه پالیدال برای درمان تورتیکولی در مراحل ابتدایی است.
دیستونی کرامپ نویسندگان
اختلال در نوشتن یا انجام کارهایی مثل تایپ کردن و نواختن آلات موسیقی ممکن است به دلایل مختلف ایجاد شود. بیماریهای مفصلی، سندرم تونل کارپ، اختلالات مخچه یا درگیری پیرامیدال و ترموراسانسیل از جمله مواردی هستند که سبب اختلال نوشتنمی شوند. بیماران مبتلا به دیستونی کرامپ نویسندگان دچار هیچ اختلال زمینه ای قابل تشخیصی نیستند.
هنگام نوشتن به دلیل اسپاسم های دیستونیک عضلات وضع طبیعی دست تغییر میکند و سبب اختلال بارز و در موارد شدید تر مانع ادامه نوشتن می شود. ممکن است سایراعمال ظریف دست مثل بازوبستن دگمه پیراهن، استفاده ازسویچ یا پیچ گوشتی مختل شود. کرامپ نویسندگان معمولاً در سنین میانی و وبعد از آن بروز می کند. و معمولاً با پیشرفت و درگیری نواحی دیگر همراه نیست. برخی از بیماران شروع به نوشتن با دست مخالف می کنند. ولی در حدود یک سوم از این موارد دست مقابل نیز مبتلا می شود. درمان دارویی اغلب بیاثر است. تزریق توکسین بوتولیسم در مواردی می تواند به بیماران کمک کند.
بلفارسپاسم و دیستونی اورومندیبولر
بلفاروسپاسم عبارت است از اسپاسم های دیستونیک مکررعضلات پری اکولر که سبب بسته شدن چشم ها می شود. اسپاسم ها از چند ثانیه تا چند دقیقه طول می کشند و بدفعات تکرار می شوند. گاهی شدت ودفعات اسپاسم ها بحدی است که در عمل بیمار را از بینایی محروم می سازد. این اسپاسم ها اغلب با کارهایی از قبیل مطالعه، تماشای تلویزیون، یا با نور شدید تشدید می شود. دیستونی های اورومندیبولر به علت اسپاسم های مشابه در عضلات دهان، زبان و فک ایجاد می شود.
گاهی بلفاروسپاسم و دیستونی اورومندیبولر همراه هم دیده می شوند. این حالت دیستونی کرانیال یا سندرم Meige’s نام دارد. بیماران مبتلا به دیستونی کرانیال، ممکن است دچار دیستونی سرویکال و یا کرامپ نویسندگان نیز بشوند. درمان انتخابی بلفاروسپاسم تزریق توکسین بوتولیسم است. توکسین بوتولیسم به درمان برخی از بیماران مبتلا به دیستونی اورومندیبولر کمک می کند.
بیماری ویلسون Wilson’s disease
ویلسون یک بیماری نادرارثی اتوزومال مغلوب با شیوع ۳۰-۱۷ درمیلیون است. بیماری بعلت موتاسیون در ژن مربوط به پروتئین انتقال دهنده مس، یعنیcopper transportingP-type adenosine triphosphatase ATPase روی کروموزوم ۱۳ ایجاد می شود. این اختلال سبب می شود که مس بطور طبیعی از طریق صفرا دفع نشود. مس در کبد و مغز تجمع پیدا میکند. مس آزاد بسیارسمی است و سبب آسیب برگشت ناپذیر و مرگ سلولی می شود. تقریباً ۴۰ درصد موارد بیماری با علائم نورولوژیک شروع می شود. علائم بیماری بین ۴۰- ۵ سالگی بروز میکند. بروز علائم نورولوژیک بعد از ۴۰ سالگی غیر شایع است.
درگیری نورولوژیک ممکن است سیر کند یا سیر متناوب همراه با دوره های حاد تشدید علائم یا سیرسریع داشته باشد. نحوه بروز و ترکیب علائم گاهی تابلوی بالینی خاصی را بوجود می آورد. البته در بسیاری از موارد تداخل بین تابلوهای بالینی و تنوع در بروزعلائم قرار دادن بیمار را در یک گروه خاص غیرممکن میسازد. بطور کلاسیک سه تیپ درگیری نورولوژیک در بیماری ویلسون دیده می شود.
۱) نوع دیستونیک با دیزآرتری دیستونیک و د یسفاژی دیستونیک بروز میکند. ریزش بزاق از دهان drooling بعلت دیستونی عضلات ناحیه بولبر و صورت اتفاق می افتد. دیستونی عضلات صورت شکل زهر خند بخصوصی را بوجود می اورد که به risus sardonicus معروف است. علائم دیستونی دراندامها بصورت تغییر وضعیت و postureغیرعادی اندامها بروز می کند. راه رفتن حالت دیستونیک پیدا میکند و نهایتاً بیمار در وضعیت کاملاً بی حرکت بدون قدرت بلع و تکلم باقی می ماند. دراین بیماران ترمورpostural و ترمور action شایع است. ترمور میتواند دیستال یا پروگزیمال باشد. در فرم شدید و پروگزیمال، ترمورحالت شبیه بال زدن را ایجاد میکند (wing-beating). بندرت ممکن است ترمور زبان درابتدای بیماری تنها یافته غیر طبیعی باشد.
۲) فرم آکینتیک- رژید با درجات متغیر برادی کینزی و رژیدیته و صورت ماسکه و بی حالت دیده می شود. دراین فرم ترمور rest و ترمور postural شایع است.
۳) فرم پسودواسکلروتیک یا مخچه ای بیماری که بعلت شباهت ظاهری سطحی که با علائم بیماری مالتیپل اسکلروز دارد ا ینطور نامگذاری شده. دراین فرم علائم بارز در گیری مخچه ای وجود دارد که بصورت آتاکسی gait، دیزآرتری آتاکسیک واز بین رفتن هماهنگی حرکات بروز میکند. دراین فرم ترمور intentional یا آتاکسیک علامت بارزی است. شکل شدید آن به صورت ترمورهولمز دیده می شود.
از سایر علائم نورولوژیک که ممکن است در بیماری ویلسون دیده شوند، کره غیر شایع است واحتمالاً ممکن است اکثر موارد گزارش شده مربوط به مصرف داروهای آنتی کلینرژیک در زمینه دیستونی باشد. وجود تیک و میوکلونوس بسیارغیرمعمول است. تشنج تا ۶ درصد موارد گزارش شده است و در سنین پایینتر بیشتر دیده می شود. علائم درگیری پیرامیدال و علائم در گیری نوروموسکولار محیطی، اختلال حسی، واختلال اسفنکتری واتونوم در بیماری ویلسون غیر معمول است.
در حدود ۲۰ درصد موارد بیماری با علائم اختلال رفتاری و نوروسایکیاتری آغازمی شود. اغلب بیماران ۱۰۰-۳۰ درصد در گزارشات مختلف در سیر بیماری نهایتاً دچار علائم اختلال رفتاری و نوروسایکیاتری می شوند. اختلالات نوروسایکیاتری بیماری ویلسون را میتوان در پنج گروه طبقه بندی کرد. ۱- اختلالات رفتاری- شخصیتی ۲- اختلالات خلقی ۳- اختلالات شناختی ۴- اختلالات سایکوتیک ۵- سایراختلالات.
شایعترین عارضه کبدی ویلسون سیروز پست نکروتیک است. عارضه کبدی میتواند بصورت هپاتیت حاد گذرا، هپاتیت فولمینا نت، هپاتیت مزمن فعال ویا فقط هپاتومگالی آسمپتوماتیک باشد.
مهمترین علائم چشمی در ویلسون حلقه بسیار تیپیک کایزر- فلیشر Kayser-Fleischer است که به علت رسوب مس در Descemets-membrane قرنیه ایجاد می شود. حلقه KF همیشه در هردو چشم ایجاد می شود و رنگ آن از طلائی یا قهوه ای تا سبز متغیراست. روی دید اثری ندارد. ابتدا در حاشیه فوقانی قرنیه تشکیل می شود.برای دیدن آن بخصوص درافرادی که چشمان قهوه ای و مشکی دارند باید از slit-lamp استفاده کرد. موارد فوق العاده نادری گزارش شده که معدودی از بیماران مبتلا به ویلسون فاقد حلقه KF بوده اند. در بیماران ویلسون با تظاهرات غیر نورولوژیک وجود حلقهKF ارزش کمتری دارد. در فرم کبدی ویلسون ممکن است حلقه KF ایجاد نشود. علاوه براین، حلقه KF در چند بیماری کبدی دیگرمانند سیروز صفراوی اولیه نیز گزارش شده است. عارضه دیگر ویلسون در چشم کاتاراکت به شکل گل آفتاب گردان است که معمولا در دید چشم اختلال ایجاد نمیکند و در معاینه با slit-lamp در حدود ۱۷ درصد بیماران درمان نشده دیده می شود.
بیماران مبتلا به ویلسون دچار عوارض دیگری شامل ؛ آنمی همولیتیک، اختلالات خونریزی دهنده، مشکلات کلیوی، استئومالاسی و ریکتز می شوند.
درتشخیص آزمایشگاهی در ۹۵ درصد موارد کاهش سرولوپلاسمین سرم که پروتئین حمل کننده مس است مشاهده می شود. هنوز اهمیت پاتوفیزیولوژیک کمبود سرولوپلاسمین در ویلسون روشن نیست. دفع ۲۴ ساعته مس ادرار نیز افزایش نشان می دهد. اندازه گیری سطح سرمی مس ارزشی در تشخیص ندارد و میتواند گمراه کننده باشد. مصرف قرص های ضد بارداری، حاملگی و بیماری کلستاتیک کبدی سبب افزایش سطح سرولوپلاسمین سرم می شوند. برخی افراد با ژن هتروزیگوت ویلسون کاهش سرولوپلاسمین را نیز نشان می دهند. ممکن است در بیماری های کلیوی دفع ادراری مس نیز افزایش پیدا کند. دراغلب بیماران اندازه گیری سرولوپلاسمین و معاینه حلقه KF برای رد یا تایید تشخیص کفایت میکند. در موارد مشکوک انجام بیوپسی کبدواندازه گیری میزان مس موجود در بافت کبد به رد یا تایید تشخیص کمک میکند. در مواردی که انجام بیوپسی کبد ممکن نبا شد بررسی با مس رادیواکتیو ۶۴Cuغیرطبیعی خواهد بود. تعداد موتاسیون های مسئول بیماری روی کروموزوم ۱۳ بسیار زیاد است بنابراین انجام تست ژنتیک بعنوان یک روش تشخیصی عملاً امکان پذیر نیست. مگراینکه موتاسیون بخصوصی در یک جمعیت بخصوص شایع باشد.
CT اسکن یا MRI مغز آتروفی کورتیکال یا تغییرات سیگنال در ماده سفید فرونتال و در گانگلیون های بازال بخصوص در پوتامن، وضایعات تالاموس و ساقه مغز را نشان میدهد. تغییراتی از جمله علامت صورت پاندا شرح داده شده که الگوی ثابتی نیست و نمیتوان برای تشخیص بیماری ویلسون به آن تکیه کرد.
در درمان ویلسون هدف کاهش مس بدن است. سه روش برای رسیدن به این هدف وجود دارد.
۱) رعایت رژیم غذائی با حداقل مس ممکن و حذف مواد غذائی حاوی مس زیاد مثل شکلات، قارچ، صدف و جگر.
۲) استفاده از داروهایی مثل ترکیبات روی (Zn) و داروی تترا تیومولیبدات(tetrathiomolybdate) که جلوی جذب مس را در دستگاه گوارش می گیرند.
ازترکیب خوراکی سولفات یا استات روی استفاده می شود. روی اضافی در دستگاه گوارش سبب افزایش تولید پروتئین متالوتیونئین metallothionein می شود تا با روی اضافی باند شده وجلوی جذب آن را بگیرد و بالا نس فیزیولوژیک روی را حفظ نماید. متالوتیونئین با مس نیز باند می شود و در نتیجه جذب مس نیز کاهش می یابد. ترکیبات روی سبب تحریک مخاط معده می شوند. از سایرعوارض آنمی سیدروبلاستیک قابل ذکراست. افزایش آمیلاز والکالن فسفاتاز سرم و کاهش کلسترول ممکن است دیده شود. ترکیبات روی را نباید همزمان با پنی سیلامین تجویز کرد.
تترا تیومولیبدات داروی جدیدی در درمان ویلسون محسوب می شود. در دستگاه گوارش با مس والبومین باند شده جلوی جذب مس را می گیرد. تترا تیومولیبدات اثر مشابهی را پس از جذب در بافت ها نیز اعمال میکند و با ایجاد کمپلکس با البومین ومس در عمل مس آزاد را به تله انداخته، مانع اثر توکسیک آن بر سلول ها می شود. مهار برگشت پذیر مغز استخوان بدنبال مصرف تترا تیومولیبدات دیده شده است که ممکن است به دلیل اختلال دراریتروپوئز بدلیل کاهش مس در مغز استخوان باشد. از تترا تیومولیبدات بصورت یکدوره ۸ هفته ای در شروع درمان استفاده می شود. سپس بیمار را روی درمان نگهدارنده با ترکیبات روی قرار می دهند.
۳) استفاده از داروهایی که سبب تخلیه مس بدن می شوند مانند دو داروی پنی سیلامین و تراینتین (Trientin).
پنی سیلامین مؤثرترین دارو در درمان ویلسون است وهنوزبعنوان داروی اصلی درشروع درمان ودردرمان نگهدارنده ویلسون مورد استفاده قرار می گیرد. پنی سیلامین با باند شدن به مس آن را از بافت ها گرفته وکمپلکس مس- پنی سیلامین درادراردفعمی شود. در حدود دو سوم بیماران به درمان با پنی سیلامین پاسخ می دهند ومابقی علیرغم درمان با ناتوانی شدید باقی می مانند ویا فوت میکنند. علائم بهبود معمولاً پس از چند ماه و گاهی در عرض چند هفته ممکن است دیده شود. یکی از مشکلات عمده دراستفاده از پنی سیلامین وجود عوارض متعدد و جدی دارویی است. واکنش حساسیتی حاد، آگرانولوسیتوز، ضایعات پوستی، سندرم نفروتیک، سندرم گودپاسچرGoodpastures-syndrome، لوپوس، و میاستنی گراواز جمله عوارض مهم پنی سیلامین هستند. در برخی از بیماران در شروع درمان تشدید علائم نورولوژیک مشاهدهمی شود که اغلب یک پدیده موقتی است.
تراینتین (Trientin) مکانیسم اثر مشابه داروی پنی سیلامین دارد ولیاثربخشی آن کمتر از پنی- سیلامین می باشد. اغلب بعنوان جایگزین پنی سیلامین از آن استفاده می شود. اثرات جانبی کمتری دارد که شامل کولیت، تورم دئودنوم، آنمی سیدروبلاستیک، ونفریت لوپوسی است. همانند پنی سیلامین یک دوره تشدید علائم نورولوژیک در شروع درمان با تراینتین ممکن است دیده شود.
درمانهای دارویی باید برای تمام مدت عمر بیمارادامه داده شود. مواردی که درگیری شدید کبدی داشته اند با پیوند کبد درمان شده اند که سبب برگشت برخی از علائم نورولوژیک بیماری نیز شده است.
اختلالات حرکتی دارویی Drug induced movement disorders
پارکینسونیسم دارویی Drug-induced parkinsonism
شایعترین علت پارکینسونیسم دارویی نورولپتیک ها هستند. ۱۵-۱۰ در صد بیمارانی که نورولپتیک دریافت میکنند، علائم بارز پارکینسونیسم را نشان می دهند. شیوع علائم پارکینسونیسم بطور کلی با مصرف نورولپتیک ها در مدت ۳ ماه تا حدود ۹۰ درصد نیز گزارش شده است. بروزعلائم حتی با مقادیراندک داروی نورولپتیک درعرض چند روز نسبتاً شایع است. اولین و شایعترین علامت برادی کینزی است که در بسیاری اوقات تنها علامت است. آکینزی، رژیدیته، postural-instability ناپایداری وضعیت وتعادل وترمورrest وترمور چانه (rabbit syndrome) وترمور postural نیز دیده می شود. postural instability از همان ابتدا و همزمان با سایر علائم پارکینسونیسم بروز میکند. مجموعه این علائم با یک سیر حاد یا تحت حاد و بصورت دو طرفه و قرینه، ظاهر نسبتاً متمایزی با بیماری پارکینسون به سندرم پارکینسونیسم دارویی می دهد. برخلاف بیماری پارکینسون آنتی کلینرژیک ها در پارکینسونیسم دارویی بسیار مؤثرند. سایر داروهائی که ایجاد پارکینسونیسم دارویی میکنند عبارتند از:
-رزرپین و تترابنازین که با تخلیه ذخایر دوپامین پره سیناپسی در واقعبصورت آنتاگونیست های پره سیناپتیک دوپامین عمل میکنند.
-بلوکر های کلسیم مثل فلوناریزین و سیناریزین که در درمان میگرن، ورتیگو و tinnitus استفاده می شود.
-دارو هائی مثل لیتیوم، والپروآت سدیم و بندرت داروهای SSRI
علائم پارکینسونیسم دارویی معمولاً پس از قطع داروی ایجاد کننده علائم درمدت چند هفته تا چند ماه از بین می رود. در صورت ادامه علائم احتملاً بیماری پارکینسون زمینه ای از قبل وجود داشته است.
واکنش دیستونیک حاد Acute dystonic reaction
بصورت اسپاسم حاد فک، دهان، زبان یا گردن وسایر اندامها و گاهی همراه باانحراف چشم ها (کریز (Oculogyric بفالصه ۲۴ تا ۴۸ ساعت پس از مصرف داروی نورولپتیک در ۲ تا ۵ درصد افراد اتفاق می افتد. نوجوانان و مردان جوان برای واکنش دیستونیک حاد ریسک بیشتری دارند.
حملات دیسترس زیادی برای بیمار ایجاد میکند. دزمان انتخابی تزریق وریدی آنتی کلینرژیک ها است.
آکاتژیا Akathisia
احساس بیقراری سایکوموتور است. فرد مبتلا مجبور به راه رفتن و حرکت مداوم می شود تا آرامش نسبی پیدا کند. در حالت نشسته مرتباً پاها و دستها را حرکت می دهد. آکاتژیا با پارکینسونیسم دارویی قرابت دارد و حتی در پارکینسون ایدیوپاتیک نیز دیده می شود. ممکن است بخوبی به درمان پاسخ ندهد. از داروهای آنتی کلینرژیک، پروپرانولول و کلونازپام در درمان آن استفاده می شود.
دیس کینزی های تاردیو Tardive dyskinesias
حرکات غیر طبیعی غیر ارادی هستند که بعد از چند ماه یا چند سال درمان با نورولپتیک ها (ضمن درمان یا پس از کم کردن مقداردارو) در بیماران ظاهر می شوند.
شایعترین فرم آن دیس کینزی کره ای فرم اوروفاشیال است که بصورت حرکات تکرار شونده لبها، دهان و زبان تظاهرمی کند. شیوع آن در زنها بعد از میانسالی بیشتر است. در حدود ۶۰ درصد موارد با قطع نورولپتیک بتدریج بهبود می یابد.
در بیماران جوانتر دیس کینزی تاردیو ممکن است بصورت سندرم دیستونیک بروز نماید. که غلب با رتروکولیس همراه با درگیری سایر اندامها تظاهر می کند. متاسفانه فقط ۱۵ درصد بیماران شانس بهبودی دارند.
هرگاه ادامه مصرف داروهای نورولپتیک بدلیل بیماری روانپزشکی زمینه ای ضروری باشد بهتر است از داروهای نورولپتیک آتیپیک استفاده شود.
دیس کینزی تاردیو اوروفاشیال به تترابنازین پاسخ می دهد ولی با آنتی کلینرژیک ها بدتر می شود. دیستونی تاردیو با آنتی کلینرژیک بهتر می شود.
سندرم نورولپتیک بدخیم Neuroleptic malignant syndrome
انواع داروهای بلوک کننده رسپتورهای دوپامین ممکن است سبب بروز علائم بالینی بصورت رژیدیته شدید، تب، نارسایی اتونوم همراه با اختلال هوشیاری شوند. از نظر آزمایشگاهی کراتین کیناز سرم افزایش نشان می دهد و اغلب لکوسیتوز وجود دارد. درمان قطع داروی ایجاد کننده علائم و استفاده از آگونیستهای دوپامین و استفاده از شل کننده عضلانی دانترولن است.
بله میتونید با متخصص ژنتیک در این خصوص مشاوره کنید.
به هرحال در کشور دستوراتی صادر شده که با بررسی مرتب در دوره بارداری و در سونوگرافی اگر خدایی نکرده مشکل خاصی بود از تولد و رشد جلوگیری بشه.
پس در این خصوص نباید نگران باشید ولی همیشه باید درصدی رو هم قبول کنید که ممکنه اتفاقی بیفته پس با مشاوره خودتون رو از این نگرانی رها کنید
سندرم گیلن باره یا بیماریGBS
عذر می خوام اگر داروی خاصی سراغ دارید دراین زمینه راهنمایی بفرمایید
پاسخ
دوست عزیز اگر منظور شما
GBS
بوده باشه باید گفت مطالب خاصی که بیشتر از حوه علمی روز دنیا باشه در اختیار نیست
علت تاخیر هم در پاسخگویی برای جستجو و مشورت بود
سندرم گیلن باره (Guillain-Barré syndrome) اختلال نادری است که در اثر حمله سیستم ایمنی بدن به اعصاب محیطی ایجاد می شود.
به دنبال این حمله، احساس ضعف در ماهیچه ها، بی حسی، گزگز و گاهی اوقات فلجی دیده می شود.
در این بیماری سیستم ایمنی بدن به اعصاب حمله میکند که باعث ضعف بدن و مشکلات حسی میشود.
سوزش و حالت خواب رفتگی ناگهانی در پا و انتقال این حس در نقاط مختلف بدن یکی از علایم بارز این بیماری است که ممکن است با حسی در پا و همراه با ضعف عضلانی بدن رخ دهد.
این بیماری بیشتر در سنین ۳۰ تا ۴۰ سالگی دیده شده است، ولی ممکن است در هر سنی اتفاق بیفتد.
این بیماری مزمن نیست و با یک دوره دارویی قابل درمان است که در صورت درمان نشدن به موقع ممکن است مشکل تنفسی ایجاد کرده و بدن را بسیار ضعیف کند.
علل گیلن باره
علت این بیماری هنوز به روشنی مشخص نیست، ولی معمولا بعد از یک عفونت ویروسی مانند سرماخوردگی و یا آنفلوانزا اتفاق می افتد.
دلیلش میتواند این باشد که سیستم ایمنی بدن برای مبارزه با عفونت به شدت تلاش میکند و ممکن است که در این میان به صورت اشتباهی به سلولهای عصبی خودی حمله کند.
همچنین بعد از عفونت یک باکتری به نام کامپیلوباکتر هم ممکن است بیماری بروز نماید. عفونت با کامپیلوباکتر ممکن است بعد از خوردن آب آلوده یا غذای نپخته (مخصوصا مرغ و ماهی و…) رخ دهد.
بعضی موارد ممکن است علایم گیلن باره بعد از اعمال جراحی اتفاق بیفتد.
گاهی هم هیچ گونه عامل شناخته شده ای وجود ندارد، اما در همه موارد ذکر شده به طور حتم اختلال در عملکرد سیستم ایمنی بدن وجود دارد.علایم گیلن باره
نشانه های گیلن باره معمولا با بی حسی و ضعف در پاها شروع می شود (ممکن است شما احساس گزگز شدن داشته باشید).
نشانه ها اغلب در عرض مدت کمی حدود چند روز یا چند هفته به قسمت های بالایی بدن کشیده می شود و بازوها و اندام فوقانی ضعیف و بی حس می شوند.
گاهی اوقات ضعف آن قدر شدید است که بیمار اصلا قادر به راه رفتن نیست و همچنین ممکن است به فلجی کامل بدن منجر شود.
همچنین ممکن است بیمار در نفس کشیدن دچار مشکل شود.
تشخیص گیلن باره
پزشک با دیدن علائمی مثل ضعف یا مورمور شدن پاها به بیماری گیلن باره شک میکند.
بعد از آن آزمایشاتی مانند گرفتن مایع مغزی- نخاعی از قمست پشت انجام خواهد داد.
دو آزمایش دیگر هم انجام میشود که شامل بررسی الکترونیکی عضله و آزمایش سرعت هدایت عصب است. این آزمایشها برای بررسی نحوه ارسال پیغام از طریق اعصاب به دستها و پاها انجام میشوند.
درمان گیلن باره
گلین باره درمان قطعی ندارد، اما راه هایی وجود دارد که علائم را بهبود می بخشد :
الف) ایمنوگلوبولین درمانی: این دارو از جنس پروتئین است و از سیستم ایمنی در برابر حملات آنتی بادی های مهاجم محافظت می کند.
ب) پلاسما فرزیس: دستگاهی به بدن شما وصل می شود که خون به درون آن کشیده می شود و آنتی بادی های حمله کننده به اعصاب بدینوسیله از خون جدا می شوند. این آنتی بادی ها، پروتئین هایی هستند که در خون وجود دارند و باعث حمله به سیستم ایمنی بدن می شوند.
بیمار مبتلا به سندروم گیلن باره نیاز دارد که چند روز تا چند هفته در بیمارستان بماند. زمان بستری ماندن در بیمارستان به شدت بیماری و مقدار مراقبتهایی که بیمار احتیاج دارد، بستگی دارد.
در موارد پیشرفته بیماری ممکن است فرد قادر به خوردن از راه دهان نباشد که برای او لوله بینی – معدی می گذارند و از طریق لوله به او غذا داده می شود.
رژیم غذایی بیمار باید پرکالری و پر پروتئین باشد تا قدرت عضلاتی او حفظ و بازسازی شود.
ممکن است غذا با حجم کم و در دفعات زیاد برای بیمار تجویز شود.
در زمان بستری بودن در بیمارستان، اگر ضعف عضلات تنفسی موجب اختلال تنفس در فرد شود، ممکن است به دستگاه تنفس مصنوعی نیاز پیدا کند.
ممکن است برای کمک به حفظ قدرت و انعطاف پذیری عضلات، فیزیوتراپی برای فرد انجام شود.
علاوه بر موارد فوق از داروهای زیر برحسب نیاز و به منظور تسکین علایم استفاده می شود :
– کورتون که التهاب و تورم اعصاب را کاهش می دهد.
– سرکوب کننده ایمنی مانند آزارام یا سایتوکسان که در مرحله حاد بیماری تجویز می شود.
– مسکن ها و ضد درد ها
– سایر داروها مانند کاربامازپین
روند بیماری گیلن باره
این بیماری سه مرحله حاد، نقاهت و بهبودی دارد.
معمولا افراد مبتلا به گیلن باره بهبود می یابند، اما ممکن است زمان زیادی طول بکشد.
در اکثر افراد، ضعف در طی ۲ تا ۳ هفته بدتر شده و سپس شروع به بهتر شدن می کند و بعد از مدتی به طور کامل از بین می رود.
اگرچه بهبودی کامل ممکن است تا دو سال طول بکشد.
بعضی افراد تا ماه ها ضعف دارند و در موارد کمی، این ضعف تا سال ها باقی می ماند، بعضی دیگر هم هرگز قدرت عضلاتشان بر نمی گردد.
گاهی اوقات این بیماری بعد از اولین حمله دوباره عود می کند.
بعضی وقتها بیمار نیاز دارد که مدتی با صندلی چرخدار یا با کمک عصا راه برود تا این که دوباره عضلاتش قوی شده و بتواند به راحتی و بدون نیاز به کمک، قدم بردارد.
فیزیوتراپی میتواند در قوی شدن عضلات و یادگیری دوباره راه رفتن و حرکت دادن ماهیچههای بدن به بیمار کمک کند.
فیزیوتراپیست برنامهای برای تمرین و ورزش به شخص بیمار آموزش میدهد تا در به دست آوردن دوباره سلامتی به او کمک کند.
کودکی که دوره درمان سندروم گیلن باره را میگذراند، ممکن است احساس غمگینی، عصبانیت، بیهودگی یا همه اینها را داشته باشد.
مشورت با یک درمانگر یا مشاور میتواند در کنترل احساسات و خشمهای روحی به کودک کمک کند.
خانواده و دوستان هم میتوانند با حمایت و درک کودک بیمار به او کمک کرده و اسباب خوشحالی او را فراهم کنند تا این که به بهبودی کامل برسد.
نحوه مراقبت از خود
– از مصرف سیگار و الکل خودداری کنید.
– با دقت توصیه های پزشک را دنبال کنید.
– داروی خود را صحیح و سر وقت مصرف کنید.
– اگر فیزیوتراپی می شوید، به دقت ورزش های فیزیوتراپی را انجام دهید تا عضلاتتان دوباره انعطاف پذیر و قدرتمند شود.
– مشاوره با یک روانشناس به سازگاری شما با شرایط بیماری و کیفیت زندگی مطلوب کمک می کند.
مشکل محدودیت چشم چپ مادرزادی
٣۰ سالمه . و بصورت مادرزاد مشکل محدودیت چشم چپ دارم, آیا راهی برای برطرف کردن این مشکل وجود داره؟
پاسخ
با متخصص مربوطه مشورت کنید
در این مورد میتونید با متخصصین کانون مشاوران ایران، مشاوره تلفنی/تخصصی داشته باشید
۰۲۱-۲۲۳۵۴۲۸۲
پارکینسون
با سلام ، امروز با سایت شما آشنا شدم ۴۴ سال دارم که یک ساله به علت پارکینسون تحت معالجه می باشم به دلیل عواقب نگران کننده آن خواهشمند است :
۱- اگر انجمن پارکینسون در ایران هست معرفی فرمائید.اگر نیست
۲- برای اخبار جدید و مشاوره جه پیشنهادی دارید.
متشکرم
پاسخ
در این مورد میتونید با متخصصین کانون مشاوران ایران، مشاوره تلفنی/تخصصی داشته باشید
دفتر قیطریه:
۰۲۱-۲۲۶۸۹۵۵۸ خط ویژه
دفتر سعادت آباد:
۰۲۱-۲۲۳۵۴۲۸۲ خط ویژه
دفتر شریعتی:
۰۲۱-۸۸۴۲۲۴۹۵ خط ویژه
آتروفی مخچه اکتسابی
آیا آتروفی مخچه اکتسابی درمان دارد؟
پیشاپیش از توجه و پاسخگویی شما سپاسگزارم
تا به حال نام بیماری “آتروفی مخچه” به گوشتان خورده است؟
به احتمال زیاد نشنیده اید؛ حق دارید.
این بیماری از جمله بیماری های نادر است که درصد کمی از مردم دنیا به آن مبتلا هستند و متاسفانه علم پزشکی جهان نیز هنوز درمان قطعی برای این بیماری نیافته است.
به گزارش پایگاه اطلاع رسانی نادر نیوز؛ تحقیقات اخیری که در ژاپن به انجام رسیده، تاثیر مثبت بعضی از گیاهان دارویی سنتی ژاپنی را در درمان این بیماری نشان داده است اما در مجموع، در حال حاضر هیچ روش درمانی برای درمان آتروفی مخچه در دست نیست.
نشریه “سفیرسلامت” برای روشن شدن زوایای پنهان بیماری آتروفی مخچه با دکتر محمدرضا معتمد؛ متخصص بیماری های مغز و اعصاب و عضو هیات علمی دانشگاه علوم پزشکی ایران گفتگویی را ترتیب داده است که مشروح آن را در ادامه از نظر می گذرانید:
آقای دکتر به عنوان اولین سوال؛ بیماری آتروفی مخچه چیست؟
مخچه قسمتی از سیستم عصبی است که فعالیت اصلی آن هماهنگی بین کلیه حرکات بدن است؛ در حقیقت اگر کارآیی مخچه تقلیل یابد توانایی انجام کارها از بین نمی رود اما هماهنگی بین حرکات اعضای مختلف بدن مختل می شود.
به عبارت دیگر، فعالیت مخچه را می توان به یک گروه ارکستر موسیقی تشبیه کرد؛ گروهی که شامل نوازنده های قابلی است اما در این بین، اگر نتی که قرار است توسط تمامی اعضای گروه نواخته شود با هماهنگی نواخته نشود، قطعا صدای ناخوشایندی را به همراه خواهد داشت.
بنابراین فعالیت مخچه نقشی برروی حرکات بدن ندارد اما در هماهنگی و برقراری ارتباط بین کلیه سیستم های بدن از جمله لب، زبان، دندان، تارهای صوتی، حرکات دست و پا و … تاثیرگذار است؛ لذا به طور قطع در صورت رسیدن هر نوع آسیبی به مخچه، فعالیت های بدن مختل خواهد شد.
حال در این میان، مخچه ممکن است به شکل های مختلفی دچار آسیب شود از جمله انسداد رگ های خونی مرتبط با مخچه که به این بیماری، اصطلاحا سکته مخچه گفته می شود. شاید هم فرد دچار تومور یا خونریزی مخچه شود و در برخی موارد نیز بافت مغز شروع به کوچک شدن می کند که در این صورت فرد به آتروفی مخچه مبتلا شده است. بنابراین آتروفی مخچه بیماری است که اولا شیوع بالایی ندارد و ثانیا در این بیماری، به تدریج بافت های مغز شروع به آب شدن و تحلیل رفتن می کنند.
آیا آماری در خصوص تعداد مبتلایان به این بیماری در کشور موجود است؟
یکی از مهم ترین معضلات بخش سلامت در ایران، ضعف سیستم اطلاع رسانی و نبود آمار و ارقام دقیق از انواع بیماری های رایج در کشور است.
به عنوان نمونه بیماری “ام اس” یکی از رایج ترین انواع بیماری هاست که درصد بالایی از افراد جامعه را نیز درگیر خود کرده و مکررا در رسانه های گروهی از این بیماری صحبت به میان می آید؛ اما اگر از نهاد یا ارگانی بخواهیم که آمار دقیقی از میزان افراد مبتلا به این بیماری را ارئه دهد، قطعا پاسخ منطقی و صحیحی در کار نیست.
حال در خصوص بیماری آتروفی مخچه که چندان شایع هم نیست و می توان گفت که عموم مردم نیز آشنایی و اطلاع کافی از این نوع بیماری ندارند، قطعا آمار مشخصی نیز وجود نخواهد داشت.
با این وضعیت، از تمامی برنامه ریزان و دست اندرکاران امر بهداشت و سلامت کشور انتظار می رود جهت تحقق این مهم گام های موثرتری بردارند چراکه بهره مندی از یک سیستم آماری متناسب می تواند جهت برون رفت از مشکلات پیش روی بیماران مثمر ثمر باشد.
در خصوص علائم این بیماری و روش هایی که برای درمان آن به کار گرفته می شود، توضیح دهید.
بخشی از بیماری آتروفی مخچه، ژنتیکی و مادرزادی و البته در برخی موارد اکتسابی است.
نوع اکتسابی آن که بسیار هم رایج است، از مصرف بی رویه انواع نوشیدنی های الکلی و استفاده طولانی مدت از برخی داروها ناشی می شود و به تدریج باعث تحلیل عملکرد سیستم عصبی مخچه شده و آتروفی مخچه را به همراه خواهد داشت.
همچنین بیماری هایی مانند کم کاری تیروئید، دیابت و یا عارضه هایی که باعث اختلال در خون رسانی مغز و عروق مخچه می شود، در درازمدت در بروز آتروفی تاثیرگذار است.
اما نوع ژنتیکی و مادرزادی آن به این صورت است که فرد در ابتدا با یک اختلال ژنتیکی مخچه متولد شده و مخچه به مرور زمان و با افزایش سن کارآیی خود را از دست داده و به تدریج کوچک شده و تحلیل رفته است.
باید توجه داشت کوچک شدن مخچه به شکل های مختلفی می تواند بروز کند؛ در برخی موارد ممکن است بیماری در همان کودکی خود را نشان دهد و گاهی نیز علائم آن در میانسالی مشخص می شود که بستگی به نوع اختلال ژنتیکی مخچه دارد.
اختلال در تکلم، سست شدن حرکات دست و پا، عدم تعادل در راه رفتن و ضعف در انجام فعالیت های شخصی و روزانه از جمله علائم بارز بیماری آتروفی مخچه است.
با این تفاسیر، پرهیز از خوردن نوشیدنی های الکلی و خودداری از مصرف بی رویه داروها علاوه بر آنکه زمینه بروز اکتشابی این بیماری را از بین می برد، در بیماران مبتلا نیز موثر واقع می شود و به کنترل بیماری می انجامد؛ اما متاسفانه درمورد نوع ژنتیکی این نوع بیماری باید گفت هنوز هیچ راه درمانی در دنیا تشخیص داده نشده است.
چند درصد از میزان ابتلا به بیماری آتروفی مخچه ناشی از مسائل ژنتیکی و چند درصد اکتسابی است؟می توان گفت درصد بالایی از میزان ابتلا به این بیماری، ژنتیکی است. یعنی چیزی بالغ بر ۶۰ تا۷۰ درصد موارد، ژنتیکی و مادرزادی است و درصد کمی از آن اکتسابی است.
اما اینکه بیشتر در چه سنی نمایان می شود قبلا هم به آن اشاره کردم؛ یک نوع بیماری پنهانی است که بسته به فرم های مختلف آن درسنین کودکی، در ابتدای سن بلوغ و یا حتی بعد از میانسالی بروز می یابد؛ بنابراین نمی توان به طور قطع گفت که آتروفی مخچه در چه سنی نمایان می شود.
این بیماری به دلیل تحلیل سیستم عصبی مخچه، توانایی های فرد را کاهش می دهد و با ضعف عضلات همراه است. با این حساب، ورزش کردن می تواند برخی نقایص بیمار را به حداقل برساند؛ همچنین توصیه من به خانواده هایی که دارای بیماری آتروفی مخچه هستند این است که از ازدواج های فامیلی به شدت خودداری کنند چرا که باعث شیوع بسیار زیاد این نوع بیماری در خانواده ها خواهد شد.
با توجه به اینکه این بیماری در دسته بیماری های نادر قرار می گیرد، بنیاد بیماری های نادر تاکنون فعالیتی برای حمایت از مبتلایان به آتروفی مخچه صورت داده است؟
به طور کلی موسسات و نهادهای غیردولتی که بعضا عمده فعالیت آنان خیرخواهانه و جهت رفع نیازها و خواسته های قشرآسیب پذیر جامعه است، توانسته اند جایگاه پررنگ تر و بس تاثیرگذارتری نسبت به نهادها و ارگان های دولتی داشته باشند.
در این میان، بنیاد بیماری های نادر نیز جزء آن دسته از نهادهای غیردولتی است که با وجود گذشت سه سال از تاسیس این مرکز، توانسته فعالیت ها و اقدامات درخور تحسینی را برای بیماران نادر انجام دهد.
ارائه خدمات درمانی مناسب، ایجاد امکانات رفاهی و تفریحی، تحت پوشش قراردادن بیمه بیماران، رفع مشکلات اقتصادی، فراهم کردن تسهیلات سفر برای بیماران و مواردی از این دست از جمله اقدامات مهم و تاثیرگذاری است که طی سه سال اخیر از سوی این بنیاد صورت گرفته است.
همچنین وجود یک مرکز آماری قوی برای شناسایی آن دسته از مبتلایان به بیماری نادر در این بنیاد و همچنین رفع به موقع خواسته ها و نیازهای این قشر از جامعه که با یک برنامه ریزی مدون و همچنین در سایه یک مدیریت کارآمد همراه است، نقش و جایگاه این نهاد را نسبت به ارگان های دولتی متمایز ساخته و دست اندرکاران و بنیانگذاران این نهاد تاثیرگذار توانسته اند با همیاری و تلاش مضاعف، مصائب بیماران را تا حدود زیادی مرتفع سازند.
توصیه ای برای بیمارانی که به عارضه آتروفی مخچه مبتلا هستند
با توجه به اینکه درصد قابل توجهی از ابتلا به عارضه آتروفی مخچه ناشی از علل ژنتیکی است که بعضا ازطریق ازدواج های فامیلی شیوع پیدامی کند، لذا از آن دسته خانواده هایی که سابقه این بیماری در آنان وجود دارد تقاضا دارم که از انجام ازدواج های فامیلی به شدت خودداری کنند تا این عارضه در خانواده شدت بیشتری پیدانکند. همچنین از بیمارانی که به صورت اکتسابی یعنی بر اساس عدم رعایت برخی نکات بهداشتی ازجمله مصرف بی رویه الکل و نوشیدنی های غیرمجاز و نیز مصرف برخی داروها به این عارضه مبتلا شده اند؛ می خواهم در درجه اول از مصرف این مواد خودداری کنند و سپس برای رفع این بیماری و درمان قطعی آن به کلیه دستورات پزشک معالج خود تا زمان بهبود کامل عمل کنند.
متاسفانه علم پزشکی تاکنون نتوانسته درمان قطعی برای این بیماری به خصوص از نوع ژنتیکی آن دست پیداکند؛ اما توصیه ای که می توان برای آن دست از بیماران مبتلا به عارضه آتروفی مخچه داشت این است که ورزش را در طول دوره درمان فراموش نکنند چراکه این امر هم به لحاظ جسمی و مهم تر از آن به لحاظ روحی بسیار تاثیرگذار است.
به این منظور توصیه اکید بنده به بیماران این است که ورزش را در برنامه روزانه خود قرار دهند.
به امید روزی که علم پزشکی در دنیا بتواند به درمان قطعی این نوع عارضه مغزی دست پیدا کند.
بیمار نادری که بیش از ۶ سالی می شود، با بیماری “آتروفی مخچه” دست پنجه نرم می کند، می گوید: بیکاری از یک سو و گرانی داروهای مورد نیاز از سویی دیگر، مرا وادار کرده است با ویلچر کنار خیابان بایستم و جوراب بفروشم.
به گزارش “سفیر سلامت”، قاسم اشکانی؛ جوان ۳۶ ساله ای است که در آستانه ۳۰ سالگی متوجه می شود به یک نوع بیماری نادر تحت عنوان “آتروفی مخچه” مبتلا شده است. می گوید در کمال سلامت بودم که شبی خوابیدم و صبح فردای آن، دیگر نتوانستم از جایم برخیزم. متاهل است و دو فرزند دختر دارد. بیماری، دو پایش را از او گرفته و حالا از واکر و ویلچر برای راه رفتن استفاده می کند. پزشکان گفته اند، بیماری قاسم ریشه ژنتیکی دارد و به صورت پنهان در بدنش رشد کرده است.
اشکانی با شکوه از اینکه امکان دسترسی به موقع به داروهای مصرفی خود را ندارد، می گوید: داروهای من کمیاب و بعضا گران است که چون توان خرید آن را ندارم، فقط به خاطر اینکه اوضاع و احوالم وخیم تر نشود و تحرکم را از دست ندهم، دوز کمتری مصرف می کنم تا داروهایم دیرتر تمام شوند.
وی از اقدامات و ارائه خدمات بنیاد بیماری های نادر قدردانی می کند و در عین حال یادآور می شود: یکی از مشکلات بیماران نادر، عدم دسترسی به موقع به پزشکان معالج وابسته به بنیاد است که امیدوارم در آینده ای نزدیک بنیاد بتواند با گسترش تعاملات خود با پزشکان، شرایطی را فراهم کند تا ملاقات پزشک و بیمار تسهیل شود.
از اشکانی در خصوص گذران زندگی با وجود شرایط نامساعد اقتصادی کشور سوال می کنیم که پاسخ می دهد: بیکاری بر زندگی من و خانواده ام سایه انداخته و مشکلات زندگی مان را دوچندان کرده است به طوریکه با ۱۰ سال سابقه فعالیت کار در شرکتی تخصصی، حال مجبور به فروختن جوراب آن هم برروی ویلچر و در کنار خیابان هستم.
اشکانی می گوید: من برای گذران زندگی به غیر از کمک و همدلی پدر و مادرم از هیچ نهاد دیگری حمایت مالی نمی شوم و از مسئولین و دست اندرکاران می خواهم برای بیماران نادر شرایطی را فراهم کنند تا حداقل از امکانات بیمه بیکاری بهره مند شویم و برای امرار معاش زندگی، جلوی خانواده خود شرمسار نباشیم.
لرزش دست چپ/کندی حرکات
مادر من ۶۹ سال دارد و مدت ۳ ماه است دارای لرزش دست چپ/کندی حرکات و از دست دادن موردی تعادل است.متخصص مغز و اعصاب تشخیص پارکینسون داده بعد متوجه شدیم تری فلوپرازین هم ممکنه در این مورد تاثیر داشته باشه و در حال قطع آن هستیم.میخواستم بدونم مشکل ایشان پارکینسون است یا پارکینسونیسم؟ و آیا امدیدی به بهبودی و پیش گیری از گسترش بیماری هست یا نه؟
پاسخ
در مورد چنین بیماریهایی حتما” با پزشک متخصص ایشون مشورت کنید
منبع: کانون مشاوران ایران


جهش ژنتیکی در بیماری اسکولیوز